Master ABOS Board Review: Skeletal Dysplasias & Metabolic Bone Diseases | Part 2
Key Takeaway
This ABOS Board Review covers essential orthopedic bone disorders including Cleidocranial Dysplasia (CCD), Osteogenesis Imperfecta (OI), and Hyperparathyroidism. Key topics include genetic inheritance, classic diagnostic signs, skeletal and non-skeletal manifestations, radiographic findings, and management strategies for these complex conditions. Ideal for exam preparation.
Master ABOS Board Review: Skeletal Dysplasias & Metabolic Bone Diseases | Part 2
Comprehensive 100-Question Exam
00:00
Start Quiz
Question 1
A 7-year-old boy presents with a waddling gait. His mother has similar features. Physical exam reveals an unusually large head, a delayed closure of the anterior fontanelle, and the ability to bring his shoulders together anteriorly.
Which of the following genes is most likely mutated in this condition?
Explanation
Question 2
A 4-year-old child presents with severe progressive deformity of the lower extremities, profound short stature, and dentinogenesis imperfecta. Radiographs of the knees show "popcorn" calcifications at the metaphyses.
Which type of osteogenesis imperfecta does this patient most likely have?
Explanation
Question 3
A 12-year-old boy presents with a history of recurrent fractures and newly diagnosed facial nerve palsy. Radiographs reveal generalized, extreme osteosclerosis and a classic "bone-in-bone" appearance of the vertebrae. What is the primary cellular defect responsible for this condition?
Explanation
Question 4
An 8-month-old infant with a known FGFR3 mutation is brought to the clinic. The parents report recent episodes of apnea and generalized hypotonia. Which of the following is the most critical and immediate orthopedic evaluation required for this patient?
Explanation
Question 5
A 68-year-old man with a long-standing history of an enlarged skull and progressive bowing of his tibia presents with a rapidly enlarging, highly painful mass over his proximal tibia. Serum alkaline phosphatase is markedly elevated. What is the most likely diagnosis of this new mass?
Explanation
Question 6
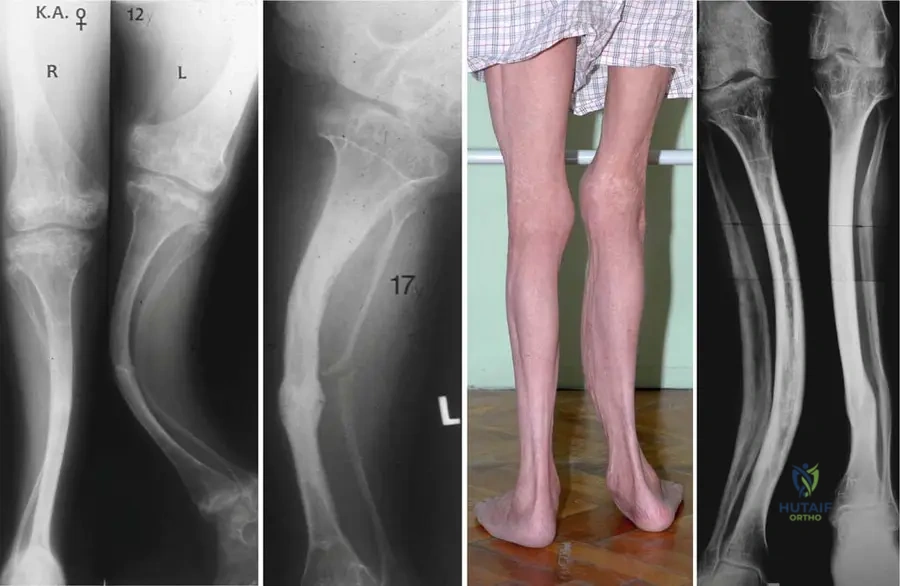
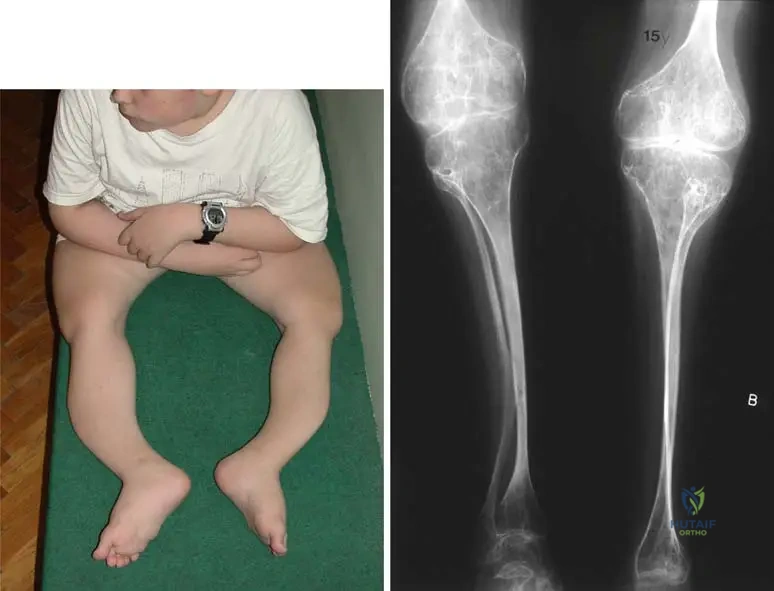
A 4-year-old boy presents with severe genu varum and short stature. Laboratory evaluation reveals normal serum calcium, low serum phosphate, normal PTH, normal 25-hydroxyvitamin D levels, and highly elevated FGF-23. What is the most appropriate medical management?
Explanation
Question 7
A 6-year-old boy presents with a short trunk, barrel chest, and coxa vara. He has normal intelligence but exhibits a waddling gait. Radiographs show flattened vertebral bodies (platyspondyly) and delayed epiphyseal ossification. Genetic testing reveals a mutation in COL2A1. Which condition is most likely?
Explanation
Question 8
A newborn is evaluated for severe bilateral, rigid clubfeet. Physical examination reveals disproportionately short limbs, "hitchhiker" thumbs, and cystic swelling of the external ear bilaterally. Which of the following genes is mutated in this condition?
Explanation
Question 9
A 10-year-old girl presents with knee pain and a waddling gait. Radiographs show small, irregular epiphyses in the hips and knees, but her spine appears radiographically normal. A lateral knee radiograph reveals a distinct double-layered patella. What is the most likely diagnosis?
Explanation
Question 10
A 2-year-old presents with bowing of the long bones, premature loss of fully rooted deciduous teeth, and a history of recurrent respiratory infections. Laboratory testing reveals strikingly low serum alkaline phosphatase levels and elevated urinary phosphoethanolamine. What is the primary deficiency in this condition?
Explanation
Question 11
A 5-year-old child presents with disproportionate short-limb dwarfism that was recognized only after he began walking. His facial features and head circumference are entirely normal. Radiographs show delayed epiphyseal ossification and anterior tongue-like projections of the vertebral bodies. Which gene is most likely mutated?
Explanation
Question 12
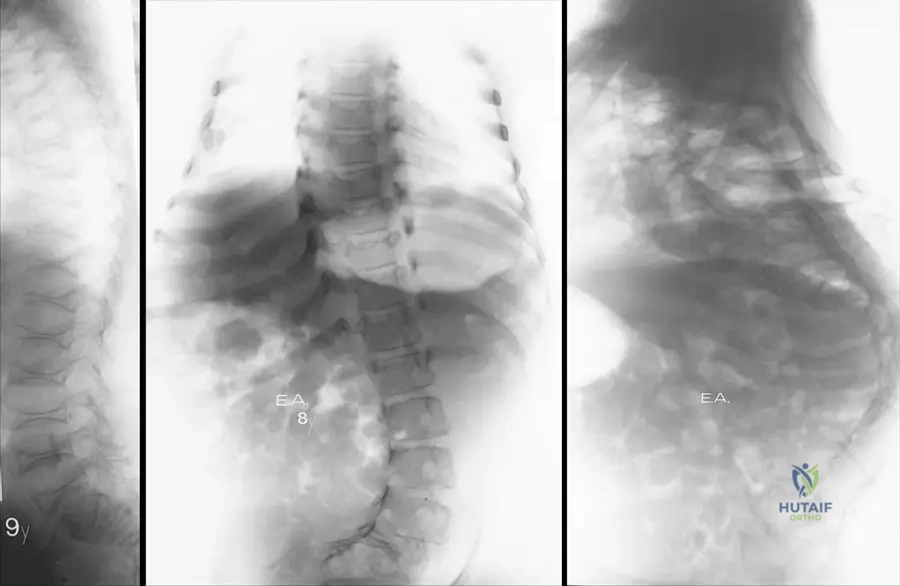
A 6-year-old boy with Osteogenesis Imperfecta presents with recurrent femoral fractures and severe progressive anterolateral bowing.
What is the surgical treatment of choice to manage the deformity and prevent fractures while allowing for continued longitudinal growth?
Explanation
Question 13
A 4-year-old boy presents with a history of recurrent fractures after minimal trauma, blue sclerae, and hearing loss.
Which of the following best describes the underlying genetic defect in the most common form of this condition?
Explanation
Question 14
A 10-year-old girl is evaluated for short stature and delayed dental eruption. On physical examination, she is able to bring her shoulders together anteriorly in the midline.
What is the genetic mutation responsible for this condition?
Explanation
Question 15
In children with severe osteogenesis imperfecta, cyclical intravenous bisphosphonates are frequently utilized to decrease fracture rates. What is the primary mechanism of action of this medication class at the cellular level?
Explanation
Question 16
A 2-year-old boy presents with generalized lower extremity pain, irritability, and bleeding gums. Radiographs show a distinct white line of Frankel and a Pelkan spur at the distal femoral metaphyses. What is the underlying biochemical defect?
Explanation
Question 17
A 6-year-old child presents with progressive genu varum, widening of the wrists, and a rachitic rosary. Laboratory studies reveal a normal serum calcium, low phosphorus, and significantly elevated alkaline phosphatase. Which of the following is the most likely initial step in the pathophysiology of this metabolic bone disease?
Explanation
Question 18
A 5-year-old boy presents with progressive bowing of the lower extremities. Laboratory tests show normal calcium, extremely low serum phosphorus, and normal PTH levels. He is diagnosed with X-linked hypophosphatemic rickets. What is the primary mediator of renal phosphate wasting in this disease?
Explanation
Question 19
An infant presents with severe anemia, cranial nerve palsies, and diffuse symmetric osteosclerosis on radiographs. A bone marrow transplant is being considered. What is the primary cellular defect causing this condition?
Explanation
Question 20
An 80-year-old man complains of increasing hat size and dull, aching pain in his right thigh. Radiographs of the femur demonstrate cortical thickening and coarse trabeculae. Which of the following best describes the histologic appearance of his femur during the mixed phase of the disease?
Explanation
Question 21
In addition to hypoplastic or absent clavicles, which of the following is a classic radiographic finding associated with cleidocranial dysplasia?
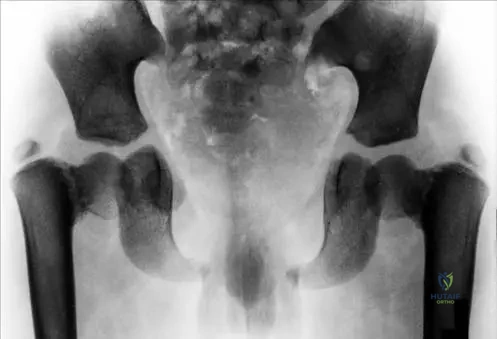
Explanation
Question 22
According to the Sillence classification for Osteogenesis Imperfecta, which type is characterized as uniformly lethal in the perinatal period?
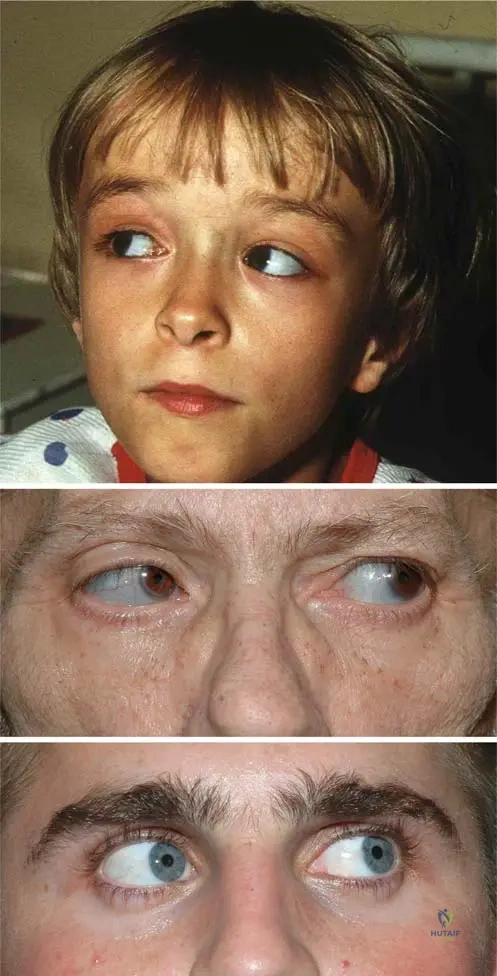
Explanation
Question 23
A 55-year-old woman with end-stage renal disease on hemodialysis presents with severe bone pain. Radiographs of her hands show subperiosteal resorption on the radial aspect of her middle phalanges. What is the primary driver of these osseous changes?
Explanation
Question 24
A neonate is diagnosed with the most common form of short-limb dwarfism. Both parents are of normal height and stature. Which of the following describes the most likely genetic mutation and its mode of inheritance?
Explanation
Question 25
A newborn presents with short stature, severe rigid clubfeet, symphalangism of the PIP joints, and "hitchhiker" thumbs. Cauliflower ears develop within the first few weeks of life. What is the underlying defect in this condition?
Explanation
Question 26
A 7-year-old boy with Sillence Type III Osteogenesis Imperfecta presents with progressive anterolateral bowing of the femur.
You elect to perform multiple osteotomies and intramedullary fixation. What is the primary biomechanical advantage of using a telescoping rod (e.g., Fassier-Duval) compared to a standard static intramedullary rod?
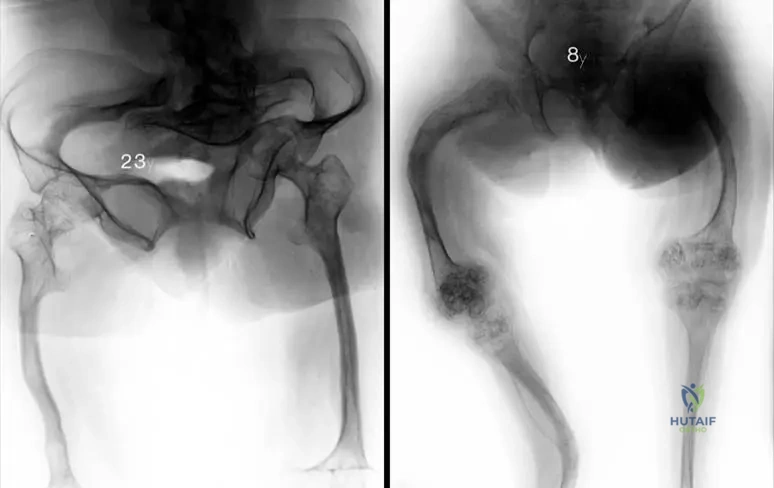
Explanation
Question 27
A 5-year-old child presents with short-trunk dwarfism, significant genu valgum, and normal intelligence. Radiographs reveal platyspondyly and hypoplasia of the odontoid process. Urine testing shows elevated keratan sulfate. Which enzyme is most likely deficient?
Explanation
Question 28
A 12-year-old girl presents with a limp. Radiographs demonstrate an expansile, ground-glass lesion in the proximal femur with a shepherd's crook deformity. She is also noted to have precocious puberty and irregular café-au-lait spots. What is the underlying molecular defect?
Explanation
Question 29
A 25-year-old patient of Ashkenazi Jewish descent presents with severe hip pain. Radiographs demonstrate bilateral avascular necrosis of the femoral heads and an Erlenmeyer flask deformity of the distal femora. Laboratory tests show pancytopenia. What is the most appropriate specific medical therapy for this underlying condition?
Explanation
Question 30
A 4-year-old boy presents with a history of recurrent fractures after minimal trauma. Clinical exam reveals blue sclerae and opalescent teeth. Radiographs show generalized osteopenia and anterior bowing of the tibiae.
What is the underlying molecular defect in this patient's condition?
Explanation
Question 31
A 7-year-old girl is evaluated for short stature and dental anomalies, including supernumerary teeth. On physical examination, she is able to bring her shoulders together anteriorly in the midline.
A mutation in which of the following genes is responsible for her presentation?
Explanation
Question 32
A 3-year-old child presents with disproportionate short stature, rhizomelic shortening, and frontal bossing. The parents are of average height. Which of the following best describes the pathogenesis of the child's most likely condition?
Explanation
Question 33
A 5-year-old boy presents with severe genu varum. Laboratory studies show normal serum calcium, critically low phosphorus, normal parathyroid hormone, and elevated alkaline phosphatase. Which of the following is the most appropriate definitive medical management?
Explanation
Question 34
A 6-year-old boy presents with a disproportionately short trunk, severe coxa vara, and an abnormal waddling gait. Cervical spine radiographs reveal odontoid hypoplasia. Genetic testing would most likely reveal a mutation affecting which of the following?
Explanation
Question 35
A newborn is noted to have severe micromelia, rigid clubfeet, and "hitchhiker" thumbs. Clinical examination also reveals prominent swelling of the bilateral pinnae. What is the underlying mechanism of this skeletal dysplasia?
Explanation
Question 36
A 14-year-old boy with Osteogenesis Imperfecta Type IV presents with new-onset hyperreflexia, lower extremity weakness, and sleep apnea.
What is the most critical and life-threatening complication responsible for these symptoms?
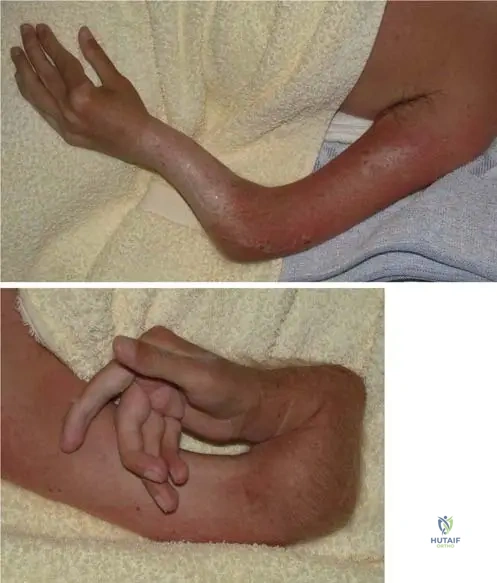
Explanation
Question 37
A 7-year-old girl with short stature, corneal clouding, and normal intelligence presents for evaluation. Radiographs show severe platyspondyly and hypoplasia of the odontoid. She is found to be deficient in which of the following enzymes?
Explanation
Question 38
A 9-year-old boy presents with hip pain and a waddling gait. Radiographs show small, fragmented, and irregular epiphyses of the proximal femora, but his spine radiographs are entirely normal. He is found to have a mutation in the MATN3 gene. Which of the following clinical findings is classic for this condition?
Explanation
Question 39
A 65-year-old man with increasing hat size and unilateral hearing loss develops sudden, severe right thigh pain. Radiographs of the femur show a new, destructive lytic lesion with cortical breakthrough in an area of pre-existing thickened, coarsened trabeculae. What is the most common malignant transformation in this patient's underlying disease?
Explanation
Question 40
A 4-year-old boy presents with severe anemia, recurrent infections, and hepatosplenomegaly. Skeletal survey reveals generalized, profound osteosclerosis with a "bone-within-a-bone" appearance in the long bones. What is the primary cellular defect responsible for this condition?
Explanation
Question 41
A 12-year-old girl is evaluated for precocious puberty and large café-au-lait spots with irregular, "coast of Maine" borders. Radiographs reveal polyostotic radiolucent bone lesions with a "ground-glass" matrix. What is the underlying genetic mutation?
Explanation
Question 42
An infant presents with failure to thrive, hypotonia, and severe bowing of all extremities. Radiographs reveal profound global demineralization and widened physes. Laboratory studies demonstrate hypercalcemia and a critically low serum alkaline phosphatase. Which gene is most likely mutated?
Explanation
Question 43
A 45-year-old patient with end-stage renal disease presents with diffuse bone pain. Radiographs demonstrate osteopenia and alternating sclerotic and lucent bands in the vertebral bodies ("rugger-jersey" spine). Which of the following sets of laboratory values is most typical for this condition?
Explanation
Question 44
A 2-year-old boy presents with anterolateral bowing of his left tibia. He has six café-au-lait spots with smooth borders on his trunk. Radiographs reveal a narrowed medullary canal and sclerosis at the apex of the bow. Which of the following is true regarding his underlying condition?
Explanation
Question 45
A 4-year-old boy presents with multiple recurrent fractures, blue sclerae, and hearing loss. Radiographs reveal generalized osteopenia and thin cortices.
What is the primary underlying biochemical defect in this condition?
Explanation
Question 46
A 7-year-old girl is evaluated for short stature and an unusual shoulder appearance. Clinical examination reveals hypermobility of the shoulders, allowing them to touch in the midline.
Which of the following genes is mutated in this patient's condition?
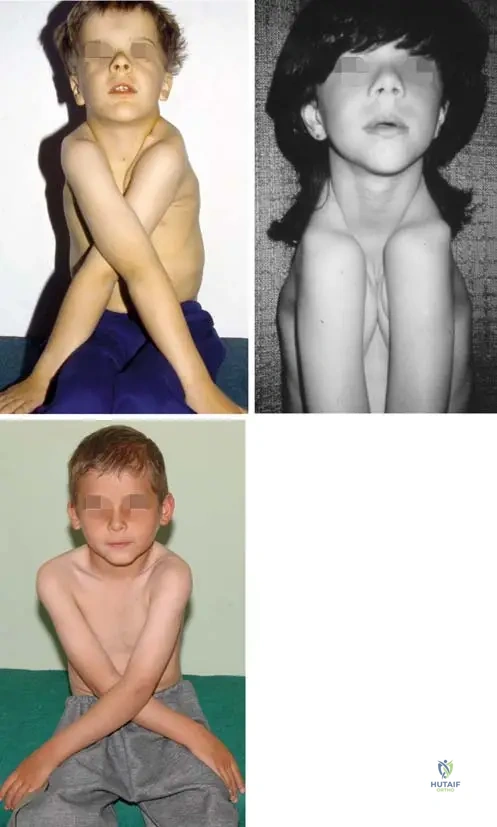
Explanation
Question 47
A 65-year-old man presents with progressive bowing of his right tibia, increasing thigh pain, and increasing hat size. Radiographs reveal cortical thickening and coarsened trabeculae of the tibia. Which of the following best describes the initial pathogenesis of this disease?
Explanation
Question 48
A 5-year-old child presents with disproportionate short stature, rhizomelic shortening, and a prominent forehead. The genetic mutation responsible for this condition primarily affects which zone of the physis?
Explanation
Question 49
A newborn presents with micromelia, bilateral clubfeet, hitchhiker thumbs, and swelling of the auricles (cauliflower ears). A defect in which of the following mechanisms is responsible for this condition?
Explanation
Question 50
A 3-year-old boy presents with progressive bowing of the legs and short stature. Laboratory evaluation shows low serum phosphate, normal calcium, normal PTH, and elevated alkaline phosphatase. Genetic testing reveals a mutation in the PHEX gene. Which of the following is the primary pathophysiologic mechanism?
Explanation
Question 51
A 45-year-old patient with end-stage renal disease complains of generalized bone pain. Laboratory results reveal elevated serum phosphorus, decreased serum calcium, and markedly elevated parathyroid hormone. Which of the following radiographic findings is most characteristic of this condition in the spine?
Explanation
Question 52
An infant is evaluated for failure to thrive, hepatosplenomegaly, and cranial nerve palsies. Radiographs show diffusely dense, bone-within-a-bone appearance in the spine. A mutation in which of the following enzymes is most commonly associated with the malignant autosomal recessive form of this disease?
Explanation
Question 53
A 10-month-old infant presents with irritability, bleeding gums, and painful swollen lower extremities. Radiographs of the knees show a dense zone of provisional calcification, a radiolucent band beneath it, and a marginal spur (Pelkan spur). Which of the following steps in collagen synthesis is primarily impaired?
Explanation
Question 54
A 9-year-old boy presents with bilateral knee and hip pain, and a waddling gait. Radiographs demonstrate delayed, irregular, and fragmented ossification centers of the femoral heads and knees, while the spine is completely normal. Mutations in which of the following genes are most commonly associated with this condition?
Explanation
Question 55
A 4-year-old boy presents with short trunk dwarfism, a barrel chest, and coxa vara. Radiographs reveal flattening of the vertebral bodies and delayed ossification of the femoral head and neck epiphyses. An abnormality in which of the following proteins is the underlying cause?
Explanation
Question 56
A 6-year-old child presents with a painful, firm mass on his back following minor trauma, and a short, malformed great toe. Over several weeks, the mass gradually turns into bone. Which of the following is the most appropriate management regarding the back mass?
Explanation
Question 57
A 6-year-old girl with osteogenesis imperfecta is treated with intravenous pamidronate.
What is the primary mechanism of action of this medication in this patient?
Explanation
Question 58
A 15-year-old boy presents for evaluation of delayed dental eruption. Examination reveals open skull sutures and absent clavicles.
Which of the following associated orthopedic conditions should also be screened for in this patient?
Explanation
Question 59
An 18-month-old child presents with enlarged wrists, bowing of the legs, and frontal bossing. Laboratory studies demonstrate low serum 25-hydroxyvitamin D, low-normal calcium, low phosphorus, and significantly elevated parathyroid hormone. Which of the following is the most likely diagnosis?
Explanation
Question 60
A 55-year-old woman presents with a pathologic fracture of her proximal humerus. Radiographs show a well-defined lytic lesion. Laboratory values reveal hypercalcemia, hypophosphatemia, and markedly elevated alkaline phosphatase. Which of the following describes the histological appearance of this bone lesion?
Explanation
Question 61
A neonate is born with severe extremity deformities, blue sclerae, and a soft skull. Radiographs show crumpled long bones, beaded ribs, and multiple fractures. The infant dies shortly after birth due to respiratory failure.
Which Sillence classification type of osteogenesis imperfecta does this patient have?
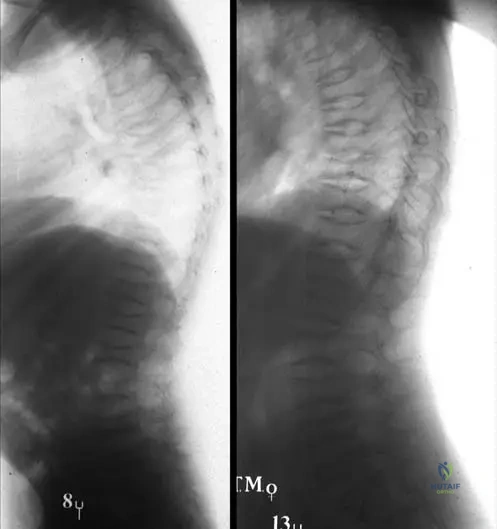
Explanation
Question 62
An 8-year-old boy presents with the ability to bring his shoulders together in the midline. Radiographs reveal hypoplastic clavicles as shown.
Which of the following genes is mutated in this condition?
Explanation
Question 63
A 10-year-old girl with delayed fontanelle closure, open skull sutures, and supernumerary teeth is evaluated for a waddling gait. What is the most common orthopedic abnormality requiring surgical intervention in this syndrome?
Explanation
Question 64
A 4-year-old child sustains a femur fracture after a minor fall. He has blue sclerae, normal dentition, and no significant hearing loss. Radiographs show generalized osteopenia.
What is the most likely Sillence classification for this patient?
Explanation
Question 65
A 12-year-old boy with a history of multiple fractures presents with progressive loss of forearm rotation. Radiographs demonstrate calcification of the interosseous membrane and a dislocated radial head. Which of the following is characteristic of his specific condition?
Explanation
Question 66
A pediatric patient with osteogenesis imperfecta is started on intravenous pamidronate. Which of the following best describes the cellular mechanism of action of this medication?
Explanation
Question 67
A 6-year-old girl with osteogenesis imperfecta type III presents with severe anterolateral bowing of the tibia.
What is the most appropriate surgical management for her tibial deformity?
Explanation
Question 68
A 2-year-old boy with frontal bossing and rhizomelic shortening of the limbs is diagnosed with a condition caused by a gain-of-function mutation in the FGFR3 gene. In which zone of the physis does this mutation exert its primary effect?
Explanation
Question 69
An 8-month-old infant with achondroplasia presents with witnessed episodes of apnea, cyanosis, and hyperreflexia. What is the most appropriate next step in evaluation?
Explanation
Question 70
A 5-year-old child presents with disproportionate short stature, normal facial features, and a waddling gait. Radiographs show delayed epiphyseal ossification and platyspondyly. A mutation in the COMP gene is identified. Which of the following cellular locations accumulates the abnormal protein?
Explanation
Question 71
A 7-year-old boy presents with bilateral hip pain and a waddling gait. Radiographs demonstrate small, fragmented proximal femoral epiphyses bilaterally and double-layered patellae. Spine radiographs are completely normal. What is the most likely diagnosis?
Explanation
Question 72
A 6-year-old child with a barrel-shaped chest, short trunk, and severe coxa vara is diagnosed with spondyloepiphyseal dysplasia congenita (SEDC). Which of the following routine screenings is critical for this patient?
Explanation
Question 73
A newborn presents with micromelic short stature, bilateral clubfeet, "hitchhiker" thumbs, and cystic swelling of the pinnae. A defect in which of the following cellular processes is responsible for this condition?
Explanation
Question 74
A 6-year-old girl with normal intelligence presents with short stature, corneal clouding, and knock knees. Urine analysis reveals excessive excretion of keratan sulfate. She is at highest risk for which of the following orthopedic emergencies?
Explanation
Question 75
A 4-year-old boy presents with progressive bowing of the legs and short stature. Laboratory tests show normal serum calcium, low serum phosphate, normal PTH, and elevated alkaline phosphatase. Genetic testing reveals a PHEX mutation. Which of the following is the primary pathophysiologic mechanism?
Explanation
Question 76
A 7-year-old child with X-linked hypophosphatemic rickets has been treated with oral phosphate and calcitriol but continues to have severe limb deformities and progressive nephrocalcinosis. What novel targeted therapy can be initiated to directly address the underlying pathophysiology?
Explanation
Question 77
An infant presents with failure to thrive, hepatosplenomegaly, and cranial nerve palsies. Radiographs show generalized osteosclerosis with a "bone-in-bone" appearance. The most severe form of this disease is characterized by a defect in which of the following?
Explanation
Question 78
A 65-year-old man presents with increasing head size, unilateral hearing loss, and progressive anterior bowing of the tibia. A biopsy of the affected bone would most likely demonstrate which of the following histologic findings?
Explanation
Question 79
A 72-year-old man with a long-standing history of polyostotic Paget disease suddenly develops severe, unrelenting pain and swelling in his right thigh. Radiographs show cortical destruction and a soft tissue mass. What is the most likely diagnosis?
Explanation
Question 80
A 55-year-old patient with end-stage renal disease presents with diffuse bone pain. Labs reveal elevated PTH, low calcium, and high phosphate. Radiographs show subperiosteal resorption of the radial aspect of the middle phalanges. What is the primary sequence of events causing this patient's elevated PTH?
Explanation
Question 81
A 6-month-old infant presents with craniosynostosis, failure to thrive, and severe rickets-like skeletal deformities. Laboratory findings are notable for significantly decreased serum alkaline phosphatase and elevated urinary phosphoethanolamine. What is the definitive treatment for this condition?
Explanation
Question 82
A 4-month-old infant with achondroplasia is brought to the clinic by her parents due to observed episodes of breath-holding and central apnea during sleep. On examination, the infant exhibits hyperreflexia in both lower extremities. What is the most appropriate next step in management?
Explanation
Question 83
A 6-year-old child presents for an orthopaedic evaluation. He has short-trunk dwarfism, a cleft palate, and severe myopia. Radiographs reveal coxa vara and flattened vertebral bodies. Prior to the patient undergoing general anesthesia for an unrelated procedure, which of the following imaging studies is mandatory?
Explanation
Question 84
A newborn is evaluated in the NICU. The infant has severely shortened limbs, bilateral rigid clubfeet, "hitchhiker" thumbs, and prominent cystic swelling of the external ears. The genetic mutation responsible for this condition primarily disrupts which of the following physiological processes?
Explanation
Question 85
A 4-year-old boy presents with progressive bowing of his lower extremities. Laboratory testing reveals normal serum calcium, low serum phosphorus, normal PTH, normal 25-OH vitamin D, and normal 1,25-OH vitamin D levels. Alkaline phosphatase is elevated. What is the primary pathophysiological mechanism underlying his condition?
Explanation
Question 86
An 8-year-old child is evaluated for disproportionate short stature and a waddling gait. He has significant joint laxity but completely normal facial features and normal intelligence. Radiographs demonstrate delayed epiphyseal ossification and irregular metaphyses. Which of the following genes is most likely mutated?
Explanation
Question 87
A 5-year-old boy presents with short stature, severe kyphoscoliosis, and corneal clouding. He also has hepatosplenomegaly. Urine analysis is highly positive for keratan sulfate. A deficiency in which of the following enzymes is responsible for this skeletal dysplasia?
Explanation
Question 88
A 12-year-old girl sustains a low-energy proximal femur fracture. Radiographs show a distinct "ground-glass" lytic lesion in the proximal femur with a shepherd's crook deformity. She has several large, irregular hyperpigmented macules on her back and a history of precocious puberty. The underlying mutation for this syndrome results in abnormal function of which of the following?
Explanation
Question 89
A 10-year-old boy of Ashkenazi Jewish descent presents with severe acute right hip pain. Radiographs demonstrate avascular necrosis of the right femoral head and a classic "Erlenmeyer flask" deformity of the distal femurs bilaterally. He also has significant splenomegaly. A defect in which of the following enzymes is the primary etiology?
Explanation
Question 90
A 14-year-old patient presents with bilateral knee pain and a waddling gait. Radiographs show flattened, irregular epiphyses at the knees and hips, and a characteristic "double-layer" appearance of the patella on the lateral view. Which of the following gene mutations is most commonly associated with this specific radiographic finding?
Explanation
Question 91
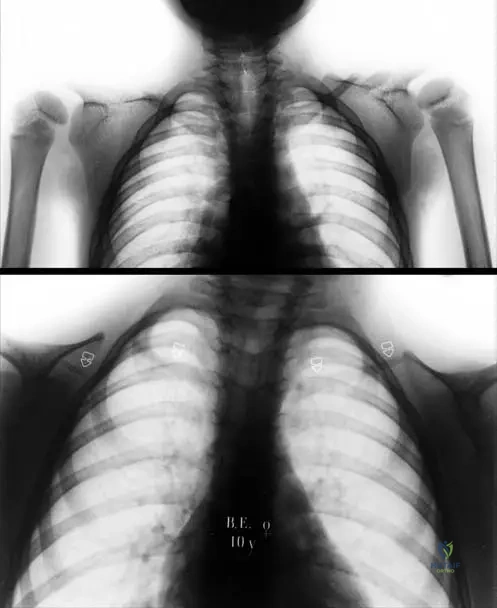
A 15-year-old girl presents for evaluation of hypermobility of the shoulders.
A radiograph of her chest and shoulders is provided. Which of the following orthopaedic conditions is most frequently associated with the underlying syndrome shown?
Explanation
Question 92
A 2-year-old girl with recurrent fragility fractures, osteopenia, and blue sclerae
is diagnosed with Osteogenesis Imperfecta and is initiated on cyclic intravenous pamidronate. What is the primary mechanism of action of this pharmacological therapy?
Explanation
None
