Master ABOS Board Review: Skeletal Dysplasias & Metabolic Bone Disease | Part 2
Key Takeaway
ABOS Bone Disorders Review covers Cleidocranial Dysplasia, Osteogenesis Imperfecta, and Hyperparathyroidism. It details their genetic patterns, classic diagnostic signs like clavicular hypoplasia or blue sclerae, skeletal manifestations such as fractures and bone resorption, and key management principles for orthopedic board exam preparation.
Master ABOS Board Review: Skeletal Dysplasias & Metabolic Bone Disease | Part 2
Comprehensive 100-Question Exam
00:00
Start Quiz
Question 1
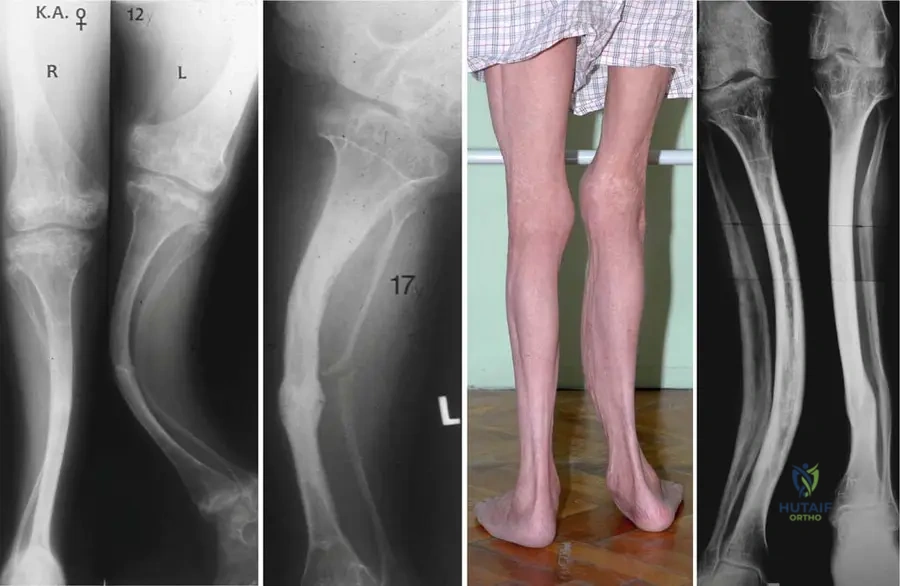
A 4-year-old child presents with recurrent fractures, blue sclerae, and the radiographic findings shown.
Which of the following genes is most likely mutated in this patient?
Explanation
Question 2
A 6-year-old boy presents with a prominent forehead, ability to appose his shoulders anteriorly, and the skull radiograph shown.
What gene is mutated in this condition?
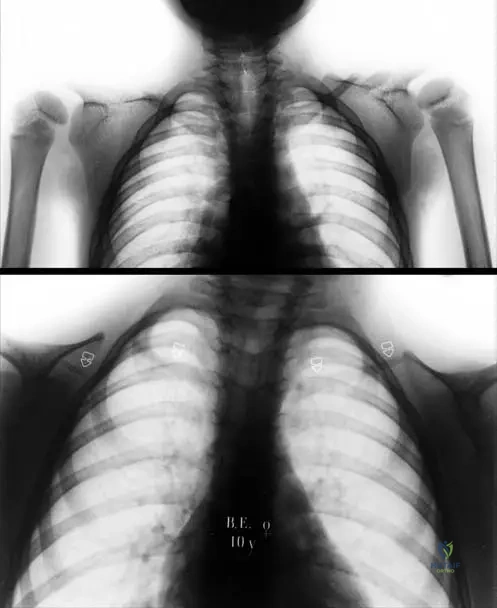
Explanation
Question 3
An infant with homozygous achondroplasia presents with progressive hypotonia, central sleep apnea, and profound hyperreflexia. What is the most appropriate next step in management?
Explanation
Question 4
A 65-year-old man presents with increasing hat size, unilateral hearing loss, and a painful, bowed tibia. Labs show an isolated, highly elevated alkaline phosphatase. Which of the following cells is the primary driver of this disease process?
Explanation
Question 5
A 5-year-old boy presents with progressive genu varum. Laboratory studies show normal calcium, low phosphorus, normal parathyroid hormone, and normal vitamin D levels. A mutation in the PHEX gene is confirmed. What is the primary mechanism of hypophosphatemia in this patient?
Explanation
Question 6
A 7-year-old girl with severe Osteogenesis Imperfecta (Type III) presents with pronounced anterolateral bowing of the bilateral femurs precluding ambulation. What is the most appropriate surgical intervention?
Explanation
Question 7
A 6-year-old child with normal intelligence presents with short-trunk dwarfism, corneal clouding, and striking knock-knees. Radiographs demonstrate platyspondyly and hypoplasia of the odontoid. What enzyme is deficient?
Explanation
Question 8
A 6-year-old boy presents with delayed closure of the cranial fontanelles, dental abnormalities, and the ability to appose his shoulders anteriorly.
Radiographs confirm the diagnosis. Which gene is most likely mutated in this patient?
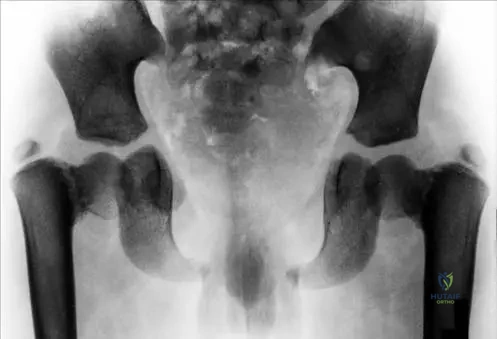
Explanation
Question 9
A 4-year-old with multiple fractures and blue sclerae is started on cyclical pamidronate therapy.
By what mechanism does this medication improve bone mineral density?
Explanation
Question 10
A newborn presents with severe anemia, cranial nerve palsies, and a diffusely dense, "bone-within-bone" appearance on radiographs. A defect in which of the following mechanisms is the primary cause?
Explanation
Question 11
A 2-year-old child with achondroplasia presents with central sleep apnea, hyperreflexia, and hypotonia. What is the most appropriate next step in management?
Explanation
Question 12
A 4-year-old boy presents with progressive bowing of the lower extremities. Laboratory evaluation reveals normal calcium, low phosphorus, elevated alkaline phosphatase, and normal parathyroid hormone levels. What is the most likely genetic mutation?
Explanation
Question 13
A 6-year-old child with a known genetic disorder presents with recurrent femoral fractures and severe progressive anterolateral bowing.
What is the most definitive surgical treatment for this long bone deformity?
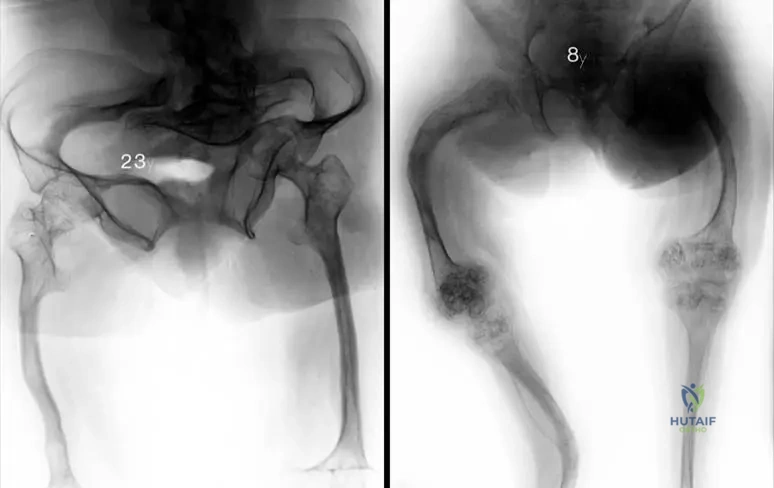
Explanation
Question 14
A 7-year-old boy presents with a waddling gait, short stature, and hip pain. Radiographs show delayed ossification of the capital femoral epiphyses and platyspondyly with normal interpedicular distances. Which protein is most likely defective?
Explanation
Question 15
A newborn presents with short-limbed dwarfism, "hitchhiker" thumbs, rigid clubfeet, and "cauliflower" ears. A mutation in which gene is responsible for this condition?
Explanation
Question 16
A 5-year-old child presents with short trunk dwarfism, corneal clouding, and severe genu valgum. Radiographs reveal platyspondyly and hypoplasia of the odontoid process. What is the most life-threatening orthopedic complication associated with this condition?
Explanation
Question 17
A 65-year-old man presents with a progressively increasing hat size, hearing loss, and a painful bowing deformity of his tibia. A bone biopsy in the initial lytic phase would most likely show which primary abnormality?
Explanation
Question 18
A 4-year-old child presents with disproportionate short stature, normal facial features, and marked joint laxity. Radiographs demonstrate delayed epiphyseal ossification and irregular, fragmented metaphyses. The underlying genetic defect involves which of the following?
Explanation
Question 19
An infant is born with a profoundly soft skull, severe long bone deformities, and multiple acute fractures.
Radiographs reveal "crumpled" long bones and beaded ribs. The infant succumbs to respiratory failure shortly after birth. Which type of Osteogenesis Imperfecta does this represent?
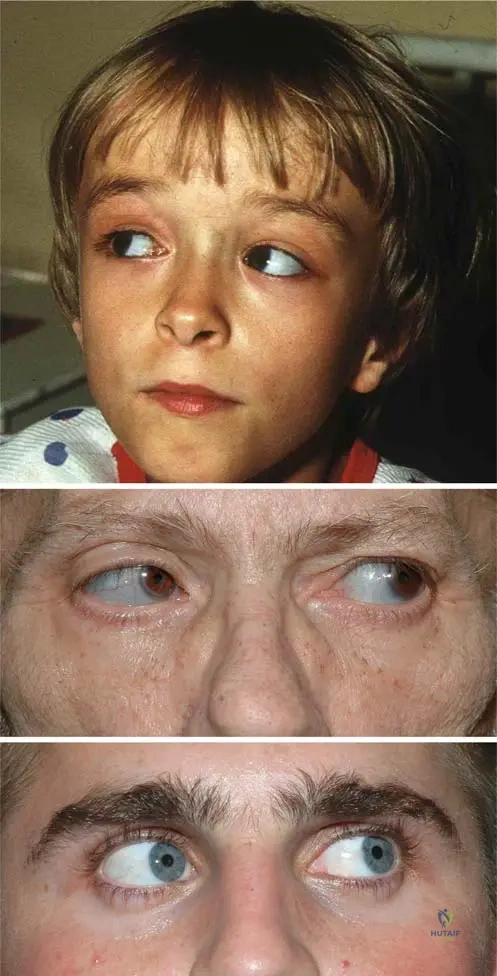
Explanation
Question 20
A 12-year-old girl with end-stage renal disease presents with bilateral hip pain. Radiographs demonstrate a "rugger jersey" spine and a slipped capital femoral epiphysis. Her PTH is markedly elevated. What is the primary mechanism driving her skeletal disease?
Explanation
Question 21
A 10-year-old girl with cleidocranial dysplasia presents with a progressively waddling gait.
Based on the typical pelvic pathology of this condition, what is the most likely diagnosis?
Explanation
Question 22
A 2-year-old boy presents with irritability, refusal to walk, and bleeding gums. Radiographs of the lower extremities show a transverse sclerotic band at the metaphysis (white line of Frankel) and a radiolucent line adjacent to it. Deficient activity of which enzyme is responsible?
Explanation
Question 23
A 6-year-old child with achondroplasia presents with progressive, symptomatic genu varum. Examination reveals fibular overgrowth relative to the tibia. What is the primary cause of this angular deformity?
Explanation
Question 24
A 3-month-old infant presents with severe bowing, recurrent fractures, and poor weight gain. Laboratory results show hypercalcemia, hypercalciuria, and markedly decreased serum alkaline phosphatase levels. Urinary phosphoethanolamine is elevated. What is the underlying molecular defect?
Explanation
Question 25
A neonate is evaluated for short-limbed dwarfism, ichthyosis, and early-onset cataracts. Radiographs demonstrate striking, stippled calcifications around the epiphyses and within the extra-articular cartilage. Which of the following conditions is classically associated with these radiographic findings?
Explanation
Question 26
A 12-year-old boy with Osteogenesis Imperfecta has been receiving long-term intravenous bisphosphonate therapy.
Radiographs demonstrate multiple transverse metaphyseal bands. Which of the following is a known potential complication of prolonged bisphosphonate use in this population?
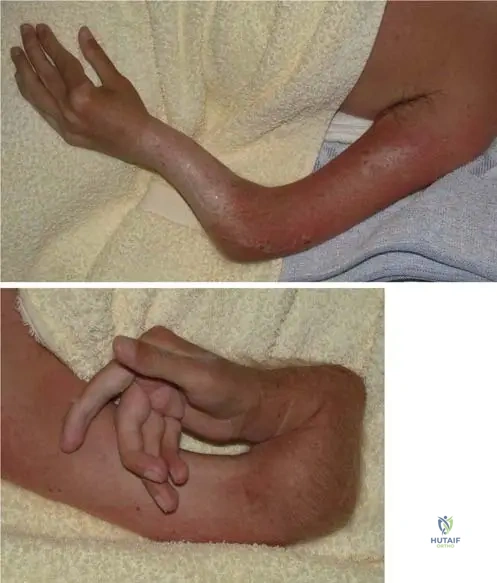
Explanation
Question 27
A 2-year-old child presents with multiple fractures, blue sclerae, and dentinogenesis imperfecta. Genetic testing reveals a quantitative defect in type I collagen. Which of the following classifications best describes this patient's condition?
Explanation
Question 28
A 7-year-old boy presents for evaluation of an abnormal gait. Radiographs show delayed ossification of the pubic symphysis, coxa vara, and absent clavicles.
This condition is caused by a mutation in which of the following transcription factors?
Explanation
Question 29
A 4-year-old girl presents with progressive genu varum. Laboratory studies show normal serum calcium, normal PTH, elevated alkaline phosphatase, and low serum phosphate. The defective gene in this condition typically leads to excessive production of which of the following?
Explanation
Question 30
A child with recurrent femur fractures is diagnosed with Osteogenesis Imperfecta and started on intravenous pamidronate. Radiographs after 2 years of treatment will most likely demonstrate which of the following findings?
Explanation
Question 31
A 6-year-old child with short-trunk dwarfism, corneal clouding, and normal intelligence develops progressive bilateral lower extremity weakness. Urine testing is positive for keratan sulfate. What is the most likely cause of the patient's neurologic decline?
Explanation
Question 32
Achondroplasia is caused by an activating mutation in the FGFR3 gene. Which specific zone of the physis is primarily inhibited by this mutation?
Explanation
Question 33
A newborn presents with micromelic shortening of the limbs, severe clubfeet, "hitchhiker" thumbs, and cystic swelling of the pinnae. Genetic testing would most likely reveal a mutation affecting which of the following processes?
Explanation
Question 34
A 5-year-old boy with Type III Osteogenesis Imperfecta presents with a highly deformed, frequently fracturing right femur.
The optimal surgical management to correct the deformity and prevent further fractures in this growing child involves which of the following?
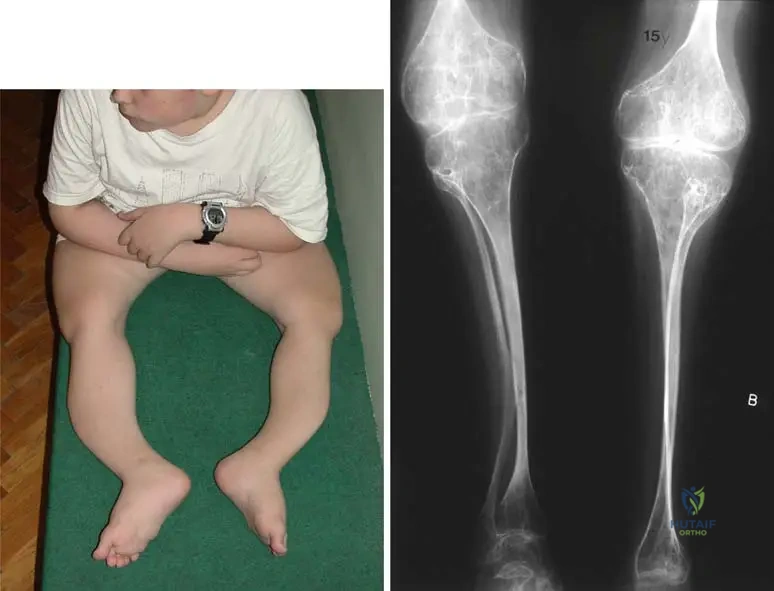
Explanation
Question 35
A 14-year-old boy with chronic kidney disease presents with bilateral hip pain. Radiographs reveal bilateral slipped capital femoral epiphyses. His serum parathyroid hormone (PTH) is markedly elevated. Which primary physiological derangement initiated this cascade?
Explanation
Question 36
A 4-year-old child is evaluated for short stature. Examination reveals short-trunk dwarfism. Radiographs show delayed ossification of the capital femoral epiphyses and coxa vara. Ophthalmologic exam reveals high myopia. The defect in this condition primarily involves which of the following?
Explanation
Question 37
A 9-year-old girl with cleidocranial dysplasia presents for follow-up.
She has bilateral coxa vara with a neck-shaft angle of 95 degrees and a Hilgenreiner epiphyseal angle (HEA) of 65 degrees. What is the most appropriate management for her hips?
Explanation
Question 38
A 7-year-old boy presents with abnormal mobility of his shoulders, allowing him to appose them anteriorly in the midline. Radiographs reveal hypoplastic clavicles and wide cranial sutures.
Which of the following best describes the underlying pathogenesis of this condition?
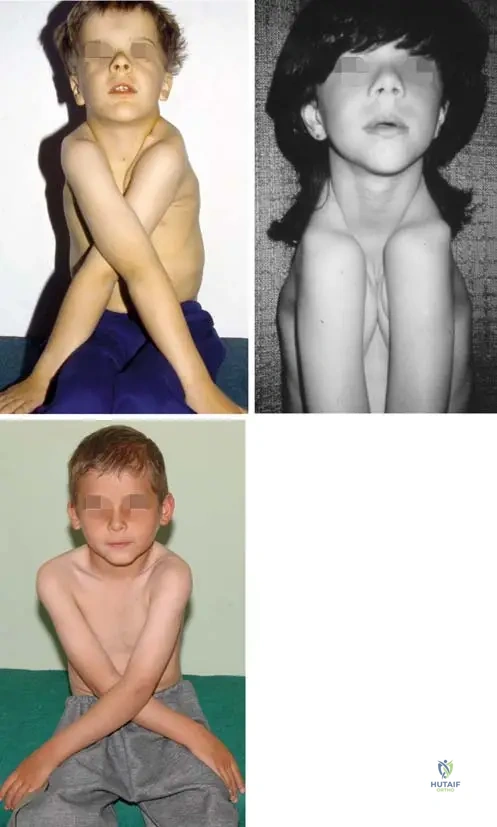
Explanation
Question 39
A 4-year-old child with a history of multiple fractures following minimal trauma presents for evaluation. Sclerae are distinctly blue.
The patient is diagnosed with Osteogenesis Imperfecta (OI). Which of the following is the most common underlying genetic mechanism for Sillence Type I OI?
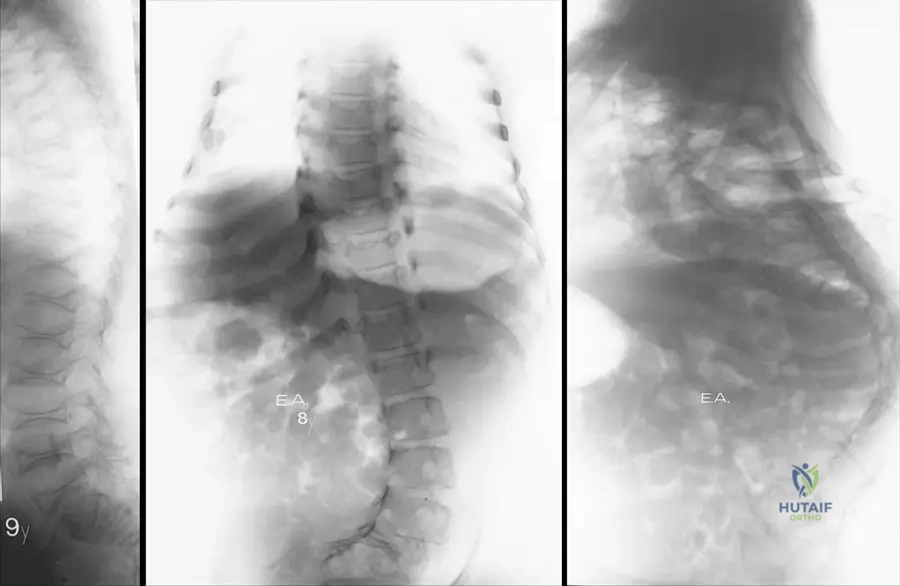
Explanation
Question 40
Which zone of the physis is primarily affected by the genetic mutation responsible for Achondroplasia?
Explanation
Question 41
A 5-year-old boy presents with progressive bowing of his lower extremities and a waddling gait. Laboratory results show normal serum calcium, markedly low serum phosphate, and elevated alkaline phosphatase. Genetic testing reveals a mutation in the PHEX gene. Which of the following is the primary mechanism leading to his bone disease?
Explanation
Question 42
A 10-year-old child presents with a history of recurrent fractures, severe anemia, and hepatosplenomegaly. Radiographs show a 'bone-within-a-bone' appearance and generalized extreme sclerosis. The primary cellular defect in this condition involves a failure to form which of the following structures?
Explanation
Question 43
A 3-year-old child presents with short trunk dwarfism, a barrel chest, and a waddling gait. Radiographs show severe coxa vara, flattened vertebral bodies, and delayed ossification of the femoral heads. Genetic analysis reveals a mutation in the COL2A1 gene. Which condition does this child most likely have?
Explanation
Question 44
Which of the following conditions is characterized by disproportionate short stature, normal craniofacial appearance, joint laxity, and is caused by a mutation in the Cartilage Oligomeric Matrix Protein (COMP) gene?
Explanation
Question 45
A 6-year-old boy with Morquio syndrome (Mucopolysaccharidosis Type IV) is scheduled for lower extremity deformity correction surgery. What critical pre-operative assessment must be performed prior to intubation?
Explanation
Question 46
A newborn presents with a 'hitchhiker's thumb', cystic swelling of the external ear, severe clubfeet, and shortened limbs. Radiographs show a shortened first metacarpal. What is the underlying genetic defect?
Explanation
Question 47
A 55-year-old patient with end-stage renal disease presents with diffuse bone pain and proximal muscle weakness. Laboratory workup reveals elevated BUN/Creatinine. Which of the following best describes the expected serum calcium, phosphate, and intact parathyroid hormone (PTH) levels in untreated renal osteodystrophy?
Explanation
Question 48
A 6-month-old infant presents with failure to thrive, hypotonia, and bowing of the long bones. Radiographs demonstrate wide, irregular physes resembling rickets. Laboratory studies reveal hypercalcemia, hypercalciuria, and markedly decreased serum alkaline phosphatase. Which substance would likely be elevated in the patient's urine?
Explanation
Question 49
A 65-year-old woman is prescribed teriparatide for severe osteoporosis. Which of the following patient history elements represents an absolute contraindication to the use of this medication?
Explanation
Question 50
An 8-year-old boy from a low-income background presents with bleeding gums, petechiae, and progressive leg pain causing him to refuse to walk. Radiographs show a dense zone of provisional calcification and a ring-shaped radiopacity around the epiphyses. The pathogenesis of this condition involves a failure of which step in collagen synthesis?
Explanation
Question 51
A 72-year-old man with known Paget's disease of the right femur presents with acutely worsening, severe right thigh pain that wakes him up at night. Radiographs demonstrate a new, destructive, lytic lesion breaking through the cortex. What is the most likely diagnosis?
Explanation
Question 52
A 10-year-old girl with severe Osteogenesis Imperfecta (Sillence Type III) develops progressive hyperreflexia, sleep apnea, and difficulty swallowing. Which of the following complications is most likely responsible for her new symptoms?
Explanation
Question 53
A 50-year-old woman presents with bone pain and a pathological fracture of the proximal humerus. Radiographs show multiple lytic bone lesions. Laboratory evaluation reveals serum calcium of 11.5 mg/dL (high), phosphate of 2.1 mg/dL (low), and markedly elevated PTH. Biopsy of a lytic lesion would most likely show which of the following?
Explanation
Question 54
A 9-year-old child presents with short stature, waddling gait, and prominent joints. Radiographs demonstrate normal vertebrae, but the patellae appear to have a 'double-layer' on the lateral view. Which of the following is the most likely diagnosis?
Explanation
Question 55
A child with Osteogenesis Imperfecta presents with progressive bowing of the femurs and a history of three femoral shaft fractures in the past year.
What is the gold standard surgical management for correcting long bone deformities in this growing child?
Explanation
Question 56
A patient with suspected Cleidocranial Dysplasia is undergoing dental and orthopedic evaluation.
In addition to clavicular hypoplasia, what is the most common dental manifestation associated with this RUNX2 mutation?
Explanation
Question 57
Which of the following phases of Paget's disease is characterized primarily by prominent, disorganized osteoblastic activity leading to woven bone formation, often manifesting radiographically as cortical thickening and bone enlargement?
Explanation
Question 58
A 4-year-old boy presents with a history of recurrent low-energy long bone fractures. Examination reveals blue sclerae and mild hearing loss. A genetic defect in which of the following is most likely responsible for this condition?
Explanation
Question 59
A 6-year-old child with severe progressive bowing of the lower extremities presents for surgical evaluation. Based on the underlying condition shown in the representative radiograph, what is the most appropriate surgical stabilization method for a femoral shaft fracture?
Explanation
Question 60
A 10-year-old boy presents with unusually prominent forehead, delayed tooth eruption, and the ability to approximate his shoulders anteriorly. Radiographs demonstrate the findings below. What is the affected gene in this disorder?
Explanation
Question 61
A 5-year-old child presents with bowing of the legs and short stature. Laboratory evaluation reveals normal serum calcium, decreased serum phosphate, normal PTH, and elevated alkaline phosphatase. Which of the following is the most appropriate medical treatment?
Explanation
Question 62
A 2-year-old boy with a known FGFR3 mutation presents with newly developed central sleep apnea, hyperreflexia, and clonus in the lower extremities. What is the most crucial next step in management?
Explanation
Question 63
An 65-year-old man presents with increasing hat size, hearing loss, and a bowing deformity of his right tibia. Which of the following best describes the initial cellular abnormality in the pathogenesis of his condition?
Explanation
Question 64
A 7-year-old child with short trunk dwarfism, corneal clouding, and normal intelligence is found to have a deficiency in N-acetylgalactosamine-6-sulfatase. Which orthopedic complication is most life-threatening in this patient?
Explanation
Question 65
An infant presents with recurrent fractures, anemia, and cranial nerve palsies. Radiographs show diffuse, uniformly dense bones with a "bone-within-a-bone" appearance. A defect in which of the following mechanisms is responsible for this condition?
Explanation
Question 66
A 55-year-old woman with end-stage renal disease on hemodialysis presents with diffuse bone pain. Lab findings reveal high PTH, low serum calcium, and high serum phosphate. Which enzyme's decreased activity in her kidneys is directly responsible for her low calcium?
Explanation
Question 67
A 4-year-old boy with a severely restricted diet of only processed foods presents with swollen, bleeding gums, perifollicular hemorrhages, and a refusal to walk due to leg pain. Radiographs reveal a dense zone of provisional calcification and a radiolucent line just proximal to the physis. Which biochemical process is defective?
Explanation
Question 68
A 6-year-old girl presents with short stature, waddling gait, and joint pain. Radiographs show fragmented, flattened epiphyses of the hips and knees, but the spine and skull are radiographically normal. A mutation in which gene is most likely responsible?
Explanation
Question 69
A neonate is born with multiple fractures, severe bowing of long bones, and a beaded appearance of the ribs. The neonate expires shortly after birth from respiratory failure. Based on the Sillence classification for this condition, what is the inheritance pattern and type?
Explanation
Question 70
A 12-year-old boy presents with progressive loss of forearm pronation and supination following a minor distal radius fracture treated in a cast. Radiographs demonstrate exuberant, hypertrophic callus formation and calcification of the interosseous membrane. Which of the following genes is most likely mutated in this patient?
Explanation
Question 71
A 7-year-old boy with a known history of Morquio syndrome (Mucopolysaccharidosis Type IV) presents for routine evaluation. His parents report he has had progressive lower extremity weakness, clumsy gait, and decreased endurance. What is the most likely structural cause of his neurologic decline?
Explanation
Question 72
An infant presents with generalized osteosclerosis, hepatosplenomegaly, and pancytopenia. Bone biopsy reveals numerous osteoclasts that completely lack a ruffled border. This form of malignant infantile osteopetrosis is most commonly caused by a mutation affecting which of the following?
Explanation
Question 73
A 4-year-old boy presents with progressive bowing of his legs. Laboratory evaluation demonstrates normal serum calcium, low serum phosphate, normal parathyroid hormone, and elevated alkaline phosphatase. Genetic testing reveals a mutation in the PHEX gene. Which of the following represents the primary pathophysiologic mechanism of this disease?
Explanation
Question 74
A 9-year-old boy presents with bilateral knee pain, a waddling gait, and difficulty running. Radiographs demonstrate delayed, irregular epiphyseal ossification and a classic "double-layered" patella on the lateral view. A mutation in which of the following genes is most commonly associated with this specific condition?
Explanation
Question 75
A 5-year-old girl presents with disproportionate short stature, joint laxity, and a waddling gait. Examination reveals perfectly normal craniofacial features. Radiographs demonstrate delayed ossification of the epiphyses and irregular metaphyses. Which of the following clearly differentiates this condition from true achondroplasia?
Explanation
Question 76
A 6-year-old child presents with short-trunk dwarfism, a barrel-shaped chest, and progressive coxa vara. Genetic testing confirms a mutation in the COL2A1 gene, consistent with Spondyloepiphyseal Dysplasia Congenita (SEDC). Which of the following extra-skeletal manifestations must be critically screened for in this patient?
Explanation
Question 77
A newborn is evaluated in the NICU and is noted to have micromelic short stature, bilateral severe clubfeet, radially deviated "hitchhiker" thumbs, and cystic swelling of the pinnae. What is the underlying genetic defect in this condition?
Explanation
Question 78
A 3-year-old girl with recurrent fractures and blue sclerae is started on an intravenous bisphosphonate protocol.
What is the primary cellular mechanism of action of this class of medication?
Explanation
Question 79
A 10-year-old girl presents for evaluation of delayed primary tooth loss and failure of permanent dental eruption. On physical exam, she is able to seamlessly appose her shoulders anteriorly in the midline.
The affected gene in this condition primarily orchestrates which of the following processes?
Explanation
Question 80
A 2-year-old child presents with markedly bowed legs, early loss of deciduous teeth with intact roots, and poor weight gain. Laboratory tests reveal hypercalcemia and unexpectedly low serum alkaline phosphatase levels. Accumulation of which of the following substances in the serum is diagnostic for this condition?
Explanation
Question 81
A 4-year-old child presents with painful, recurrent soft tissue swellings over the paraspinal muscles that progressively harden. Examination of the child's feet is most likely to reveal which of the following congenital anomalies?
Explanation
Question 82
An 18-month-old child presents with severe clinical and radiographic signs of rickets, accompanied by total body alopecia. Laboratory evaluation reveals hypocalcemia, hypophosphatemia, and markedly elevated levels of 1,25-dihydroxyvitamin D. What is the most likely diagnosis?
Explanation
Question 83
A neonate is evaluated for disproportionate dwarfism. Examination reveals a flat midface, a cleft palate, extremely short limbs, and prominent, enlarged joints. Radiographs demonstrate "dumbbell-shaped" femora and coronal clefts in the vertebral bodies. Which of the following is the most likely diagnosis?
Explanation
Question 84
A 65-year-old man presents with progressive anterior bowing of his right tibia and states his hats no longer fit. Biopsy of the affected bone reveals a mosaic pattern of lamellar bone with prominent cement lines. What is the primary initiating cellular event in the pathogenesis of this disease?
Explanation
Question 85
A newborn presents with severe anterior bowing of the lower extremities, hypoplastic scapulae, and respiratory distress due to tracheobronchomalacia. Genetic testing reveals a 46,XY karyotype, but the infant has unambiguous phenotypic female genitalia. A mutation in which gene is responsible for this condition?
Explanation
Question 86
A 7-year-old girl presents with a limp, precocious puberty, and large, irregular, hyperpigmented skin macules. Radiographs of the right femur show an expansile, radiolucent lesion with a "ground-glass" appearance. Which of the following describes the underlying genetic pathogenesis?
Explanation
Question 87
An infant presents with asymmetrical limb shortening, thick scaly skin (ichthyosis), and bilateral congenital cataracts. Radiographs reveal diffuse, stippled, calcific spots in the cartilaginous epiphyses. This presentation is most characteristic of which condition?
Explanation
Question 88
A 45-year-old patient with end-stage renal disease on hemodialysis presents with diffuse bone pain. Laboratory evaluation reveals hyperphosphatemia, hypocalcemia, and markedly elevated parathyroid hormone levels. Radiographs of the hands are most likely to show which of the following pathognomonic findings?
Explanation
Question 89
A 14-month-old child fed exclusively on boiled cow's milk presents with extreme irritability, bleeding gums, and painful, swollen lower extremities. Radiographs show a dense zone of provisional calcification (white line of Frankel) and a radiolucent band directly beneath it (Trummerfeld zone). What specific biochemical process is impaired in this patient?
Explanation
Question 90
A 5-year-old child with a known diagnosis of Osteogenesis Imperfecta presents with progressive anterolateral bowing of the femur, limiting ambulation.
What is the preferred surgical management for long-term correction of this deformity in a growing child?
Explanation
Question 91
A 7-year-old girl is evaluated for delayed eruption of secondary dentition and an unusual ability to appose her shoulders anteriorly. Radiographs confirm absent clavicles.
Which of the following orthopaedic manifestations is most commonly associated with this genetic syndrome?
Explanation
Question 92
A 6-year-old child with Morquio syndrome (Mucopolysaccharidosis Type IV) presents with progressive bilateral hand weakness, hyperreflexia in the lower extremities, and an unsteady gait. Which of the following structural abnormalities is the most likely cause of these neurologic findings?
Explanation
Question 93
A 3-year-old boy presents with progressive bilateral bowing of his legs and a waddling gait. Laboratory studies reveal normal serum calcium, markedly decreased serum phosphate, normal PTH, and elevated alkaline phosphatase. Genetic testing confirms a mutation in the PHEX gene. Which of the following best describes the underlying pathophysiology?
Explanation
Question 94
A neonate is evaluated in the NICU for multiple long bone fractures sustained during a normal vaginal delivery. Physical examination reveals soft, deformable skull bones and deep blue sclerae.
The underlying molecular defect most commonly results in a deficiency or qualitative defect of which structural component?
Explanation
Question 95
A newborn presents with short-limbed dwarfism, bilateral rigid clubfeet, "hitchhiker" thumbs, and cystic swelling of the external ears. What is the genetic inheritance pattern and affected gene for this syndrome?
Explanation
Question 96
An 18-month-old irritable infant, exclusively fed boiled cow's milk since birth, presents with gingival bleeding, lower extremity pseudoparalysis, and a prominent "white line of Frankel" on radiographs. The deficient nutrient is an essential cofactor for which of the following biochemical processes?
Explanation
Question 97
A 6-year-old child presents with short stature, a waddling gait, and joint pain. Radiographs demonstrate delayed, irregular epiphyseal ossification and normal vertebrae. The facial features are completely normal. A mutation in the COMP gene is identified. Which of the following is a classic radiographic finding of the patella in this patient's condition?
Explanation
Question 98
A 5-year-old boy with achondroplasia presents with progressive, symptomatic genu varum. Clinical examination shows significant bowing with prominent fibular heads. What is the primary anatomic etiology of the varus alignment in this condition?
Explanation
Question 99
A 3-year-old child with a history of recurrent fractures is initiated on cyclic intravenous pamidronate therapy.
What is the primary cellular mechanism of action of this medication?
Explanation
Question 100
A 1-year-old infant presents with severe bowing of the long bones, craniosynostosis, and premature loss of primary incisors. Laboratory studies reveal hypercalcemia, hypercalciuria, and a markedly decreased serum alkaline phosphatase level. Which of the following substrates is most likely elevated in this patient's urine?
Explanation
None
