Myelomeningocele: Comprehensive Orthopaedic Management and Surgical Principles
Key Takeaway
Myelomeningocele is a complex congenital neural tube defect requiring multidisciplinary care. Orthopaedic management focuses on maximizing mobility, preventing progressive spinal deformity, and addressing lower extremity contractures. Key considerations include the pathoanatomy of the everted spine, vigilant monitoring for tethered cord syndrome, and strict adherence to latex-free perioperative protocols. Surgical interventions range from spinal fusion to complex foot reconstructions.
Introduction to Myelomeningocele
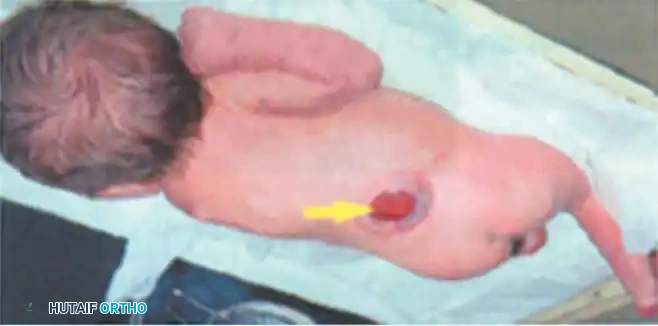
Myelomeningocele represents the most severe and complex congenital malformation of the central nervous system compatible with life, falling under the broader spectrum of spinal dysraphism and neural tube defects (NTDs). Advances in neonatal intensive care, neurosurgery, and allied health services have drastically reduced early mortality rates, shifting the clinical focus toward optimizing long-term functional independence. For the orthopaedic surgeon, the primary challenge lies in assisting these patients to attain the best possible musculoskeletal function, preventing progressive deformity, and maintaining a stable posture for either ambulation or wheelchair seating.
Spinal dysraphism encompasses a spectrum of anomalies, including meningocele, lipomeningocele, and caudal regression syndrome. A myelomeningocele specifically denotes a saclike herniation containing cerebrospinal fluid (CSF) and dysplastic neural tissue protruding through a posterior defect in the vertebral canal.
Unlike a simple meningocele—where only the cystic distention of the meninges herniates through unfused vertebral arches while the spinal cord remains within the canal—myelomeningocele involves the displacement of the spinal cord and nerve roots, resulting in variable, often profound, neurological deficits.
Embryology and Pathoanatomy
The central nervous system develops via neurulation, the formation of a tubular structure from the neural plate. Normal closure of this neural tube is completed by the fusion of the cranial and caudal neuropores between days 24 and 26 of gestation.
Two primary embryological theories exist regarding the pathogenesis of myelomeningocele:
1. Primary Failure of Closure: The neural folds fail to fuse during primary neurulation.
2. Rupture Theory: A previously closed neural tube ruptures secondary to abnormal fluid dynamics.
Regardless of the exact mechanism, the resulting pathoanatomy is profound. The myelomeningocele is formed by the protrusion of the dura and arachnoid through the deficient vertebral arches. The skin overlying the lesion is almost always absent at birth. Instead, the neural placode is covered only by a thin arachnoid membrane, which rapidly breaks down, leaving an ulcerated, granulating surface.
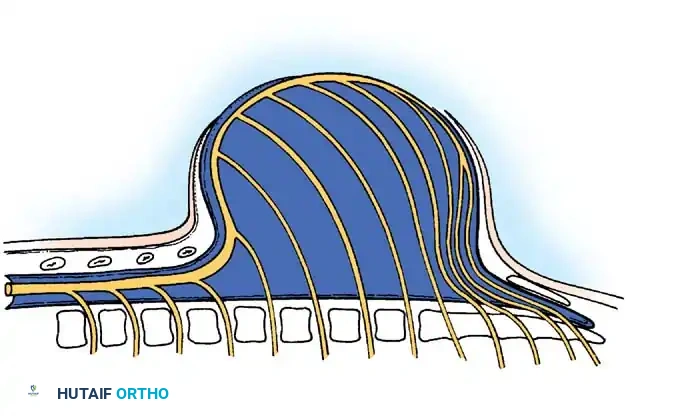
Biomechanics of the Everted Spine
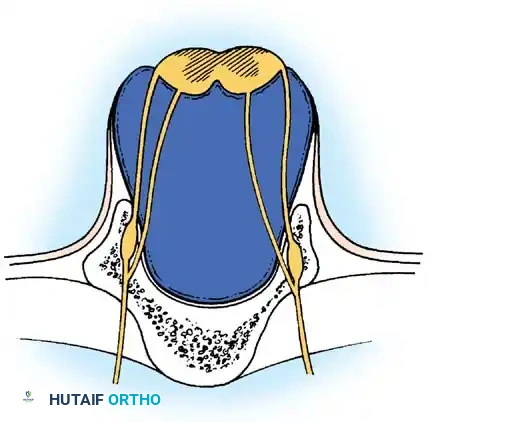
Understanding the altered biomechanics of the myelomeningocele spine is critical for surgical planning. The superficial surface of the neural placode represents the everted interior of the neural tube, while the ventral surface represents the exterior. Consequently, the nerve roots arise from the ventral aspect of the placode.
The osseous anatomy is similarly distorted. The pedicles are everted, lying almost horizontally in the coronal plane. The laminae are hypoplastic and laterally displaced. Crucially, the paraspinal muscles (erector spinae) are displaced anteriorly along with the everted pedicles. Because they now lie anterior to the axis of spinal rotation, these muscles act as paradoxical flexors of the spine rather than extensors. This relentless anterior tethering force is the primary driver of the severe, progressive congenital kyphosis seen in high-level lesions.
Epidemiology and Prevention
The incidence of myelomeningocele in the United States is approximately 0.6 to 0.9 per 1,000 live births. The true incidence of the defect is higher, but an estimated 23% of affected pregnancies are terminated following prenatal diagnosis.
The global incidence of NTDs has steadily decreased, largely due to enhanced prenatal screening and the widespread implementation of periconceptional folic acid supplementation.
Clinical Pearl: The U.S. Preventive Services Task Force (USPSTF) recommends that all women of childbearing age consume 0.4 mg of folic acid daily. For women at high risk (e.g., a previous child with an NTD or a first-degree relative with an NTD), the Centers for Disease Control and Prevention (CDC) recommends a high-dose regimen of 4.0 mg daily, beginning one month prior to conception.
Prenatal screening relies on maternal serum α-fetoprotein (MSAFP) levels measured between 16 and 18 weeks of gestation, which can detect up to 80% of affected pregnancies. Elevated MSAFP warrants high-resolution ultrasound, ultrafast fetal MRI, and potentially amniocentesis to evaluate for elevated amniotic α-fetoprotein and acetylcholinesterase.
Associated Neurological and Systemic Conditions
The natural history of myelomeningocele is heavily influenced by associated central nervous system anomalies. Orthopaedic surgeons must be acutely aware of these conditions, as they directly impact musculoskeletal function and surgical timing.
Hydrocephalus and Shunt Malfunction
Approximately 80% to 90% of children with myelomeningocele develop hydrocephalus requiring ventriculoperitoneal (VP) shunting. Before the surgical closure of the spinal defect, the ventricles communicate with the open central canal, allowing for spontaneous decompression. Once the defect is closed, hydrocephalus rapidly manifests.
Surgical Warning: Any acute or insidious change in an orthopaedic patient's neurological baseline—such as new-onset spasticity, rapidly progressive scoliosis, or loss of previously attained motor milestones—must be considered a shunt malfunction or tethered cord until proven otherwise. Orthopaedic interventions are strictly contraindicated until the neuroaxis is evaluated and stabilized by neurosurgery.
Hydrosyringomyelia
Hydrosyringomyelia is the abnormal accumulation of CSF within an enlarged central canal of the spinal cord, often secondary to altered fluid dynamics from hydrocephalus or Arnold-Chiari malformation.
Clinical manifestations include:
1. Ascending motor paralysis or increased lower extremity spasticity.
2. Rapidly progressive scoliosis.
3. Weakness or loss of fine motor control in the upper extremities and hands.
Arnold-Chiari Type II Malformation
Almost universally present in myelomeningocele, the Type II Arnold-Chiari malformation involves the caudal displacement of the cerebellar vermis and medulla oblongata through the foramen magnum into the cervical canal. This causes lower cranial nerve dysfunction, presenting as stridor, swallowing difficulties, vocal cord paralysis, and central apnea.
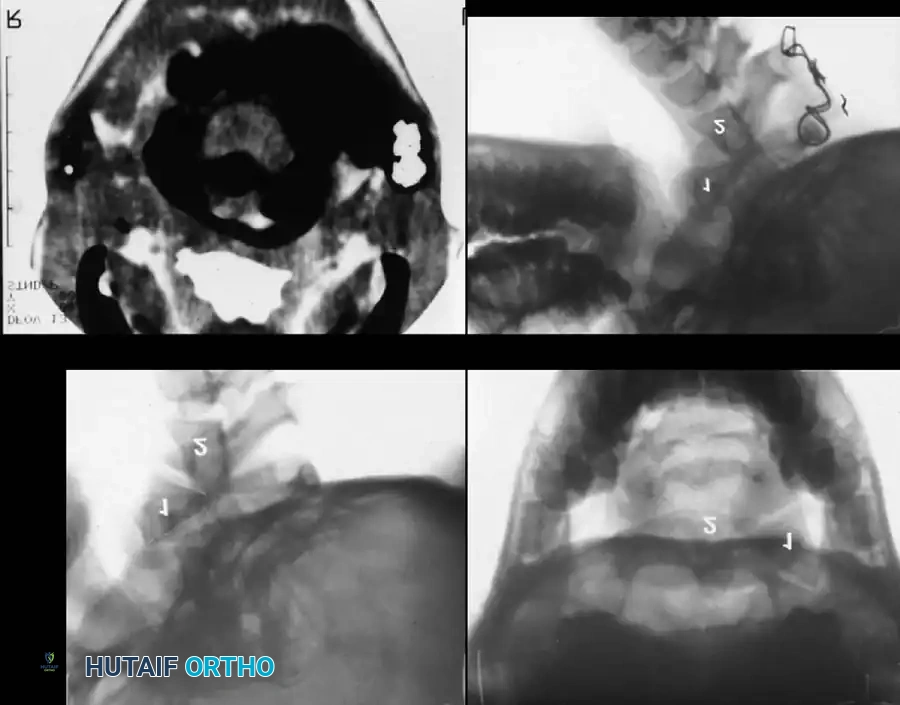
Tethered Spinal Cord Syndrome
While MRI will show anatomical tethering of the spinal cord at the surgical repair site in nearly all patients, only 20% to 30% develop clinical Tethered Cord Syndrome. The scar tissue from the initial neonatal closure prevents the normal cephalad migration of the conus medullaris during childhood growth, leading to ischemic traction injury to the cord.
Clinical signs of a tethered cord include:
* Loss of motor function or changes in manual muscle testing.
* New-onset spasticity (particularly in the medial hamstrings, ankle dorsiflexors, or evertors).
* Development of a progressive cavovarus foot deformity.
* Rapidly progressive scoliosis before age 6.
* Deterioration in urological continence.
Surgical detethering by a neurosurgeon is indicated to halt neurological decline, though it rarely restores already lost function.
Urological Dysfunction
Neurogenic bladder is present in nearly all patients. Historically, renal failure secondary to chronic pyelonephritis and hydronephrosis was the leading cause of late mortality. Modern management relies on clean intermittent catheterization (CIC) and anticholinergic medications to maintain low bladder pressures and preserve renal parenchyma.
Latex Hypersensitivity
Patients with myelomeningocele have a highly elevated risk of developing a Type I IgE-mediated latex allergy, with clinical prevalence rates around 10% and serological sensitization up to 38%. This is likely due to early and repeated mucosal exposure to latex during frequent surgeries and daily catheterizations.
Surgical Warning: A strict latex-free environment is mandatory for all patients with myelomeningocele from birth. This includes latex-free gloves, catheters, tourniquets, adhesive tapes, and anesthesia breathing circuits. Anaphylaxis in the operating room can be fatal.
Neurological Level Classification
Orthopaedic management is dictated by the patient's functional neurological level, which predicts ambulatory potential and the specific pattern of muscle imbalances.
- Thoracic Level (T12 and above): No voluntary lower extremity motor function. High risk for severe kyphoscoliosis. These patients are obligate wheelchair users.
- High Lumbar (L1-L2): Weak hip flexion (iliopsoas) and adduction present. Unopposed hip flexion leads to severe flexion contractures and hip dislocations. Wheelchair ambulators primarily.
- Mid Lumbar (L3-L4): Strong hip flexion and adduction; strong knee extension (quadriceps). Lack of hip extension (gluteus maximus) and knee flexion (hamstrings). Community ambulators with ankle-foot orthoses (AFOs) and crutches.
- Low Lumbar (L5): Weak hip extension, strong knee flexion, weak ankle dorsiflexion. Gluteus medius remains weak, resulting in a Trendelenburg gait. Community ambulators.
- Sacral Level (S1-S3): Normal hip and knee function. Weakness is confined to the intrinsic foot muscles and ankle plantarflexors (gastrocnemius-soleus). Normal community ambulators, often requiring only shoe inserts or mild bracing.
Orthopaedic Surgical Management Principles
The overarching goal of orthopaedic surgery in myelomeningocele is to provide a stable, plantigrade foot, a level pelvis, and a balanced spine centered over the pelvis.
Spinal Deformity: Scoliosis and Kyphosis
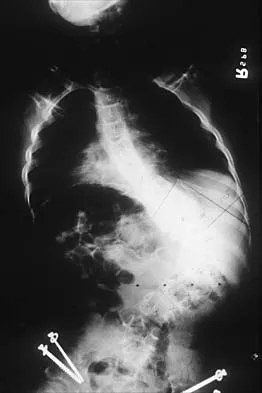
Spinal deformity is nearly ubiquitous in higher-level lesions. Congenital kyphosis is present at birth in 10% to 15% of patients, driven by the everted paraspinal muscles acting as flexors. Scoliosis develops in over 80% of thoracic-level patients.
Surgical Approach for Kyphosis (Kyphectomy):
Severe, rigid kyphosis leads to recurrent skin breakdown over the apex of the deformity, making wheelchair seating impossible. Kyphectomy is indicated for curves exceeding 80 degrees with recurrent ulceration.
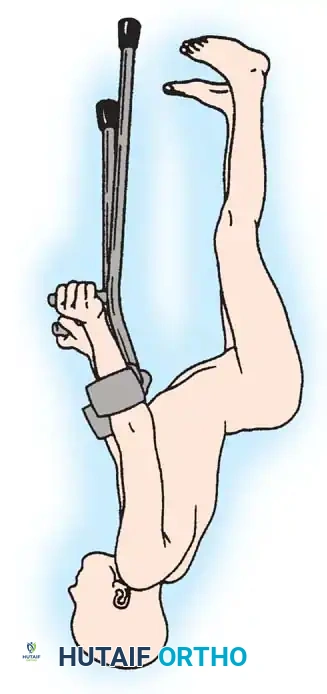
1. Positioning: Prone, with careful padding of insensate areas.
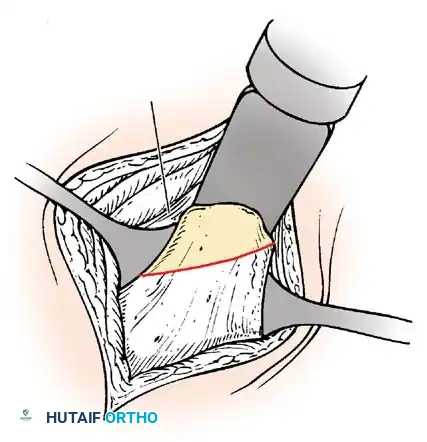
2. Exposure: A meticulous subperiosteal dissection is performed. The dysplastic dural sac is mobilized.
3. Resection: The apical 2 to 3 vertebral bodies are excised (vertebrectomy). In patients with no distal motor function, the non-functional spinal cord at the apex may be ligated and transected (with neurosurgical assistance) to allow for complete closure of the defect.
4. Fixation: Rigid segmental instrumentation is required.
Surgical Approach for Scoliosis:
Progressive paralytic scoliosis requires posterior spinal fusion, typically extending from the upper thoracic spine down to the pelvis to ensure a level seating foundation.
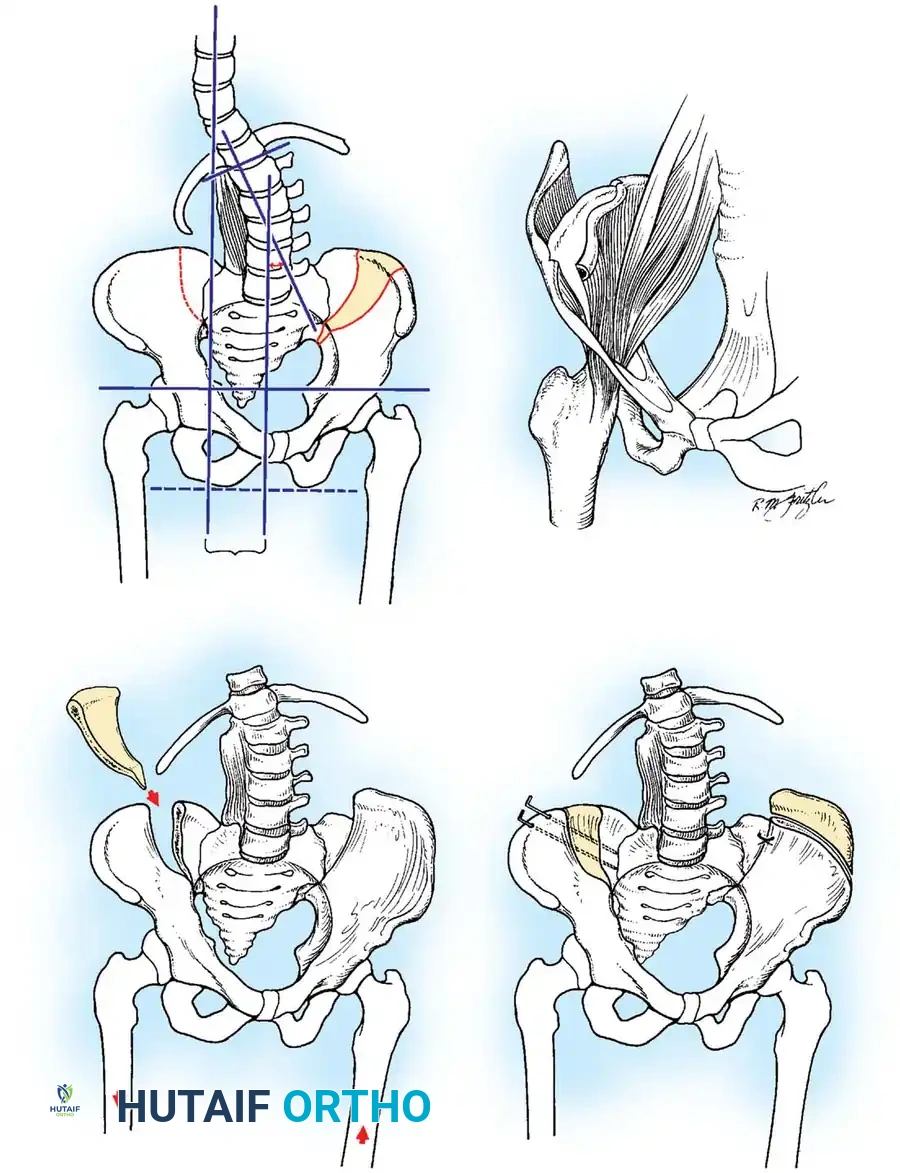
* Pelvic Fixation: Due to the deficient posterior elements (spina bifida) and poor bone quality, robust pelvic fixation is mandatory. Modern constructs utilize bilateral iliac or S2-alar-iliac (S2AI) screws.
Hip Subluxation and Dislocation
Hip instability is common, particularly in L1-L3 level patients, due to the unopposed action of the iliopsoas and adductors against paralyzed abductors and extensors.
Treatment Algorithm:
* Thoracic / High Lumbar (Non-ambulators): Bilateral hip dislocations are generally left untreated. A stiff, surgically reduced hip is detrimental to wheelchair sitting. The goal is maintaining symmetric range of motion, not radiographic reduction.
* Low Lumbar (Ambulators): Unilateral or bilateral dislocations must be reduced to maintain a level pelvis and optimize gait biomechanics.
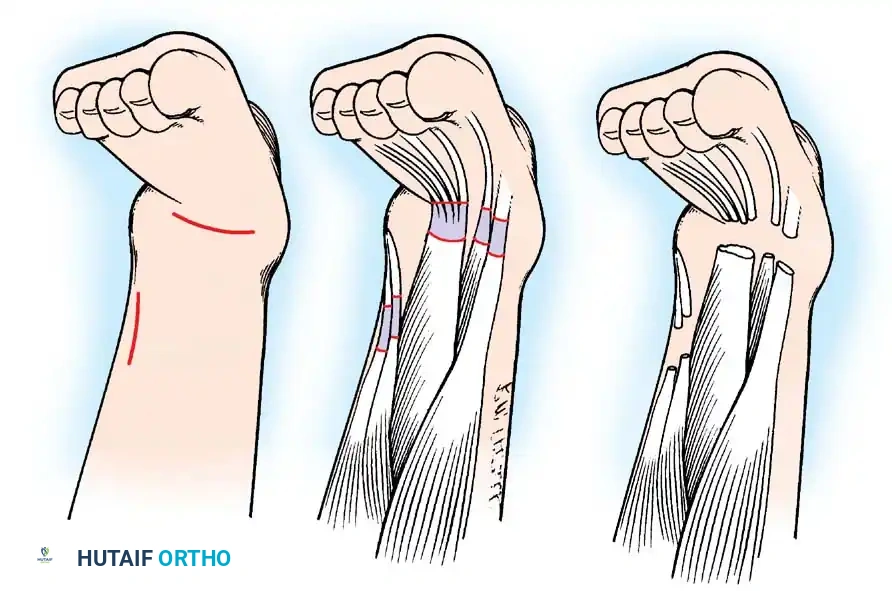
Surgical Technique (Varus Derotation Osteotomy - VDRO):
1. Soft Tissue Release: Radical release of the adductors and iliopsoas.
2. Femoral Osteotomy: A proximal femoral varus derotation osteotomy (VDRO) is performed to correct excessive femoral anteversion and coxa valga.
- Pelvic Osteotomy: If acetabular dysplasia is present, a Dega or Pemberton pelvic osteotomy is added to improve anterolateral coverage.
- Open Reduction: The hip capsule is opened, the ligamentum teres excised, and the pulvinar cleared before concentric reduction.

Knee Deformities
Knee deformities typically present as either flexion or extension contractures.
* Extension Contractures: Common in high-level lesions. Treated with V-Y quadricepsplasty if interfering with seating.
* Flexion Contractures: Common in L3-L4 lesions due to unopposed hamstrings. Contractures >20 degrees interfere with bracing and ambulation.

Surgical Management:
Mild contractures are managed with hamstring lengthening and posterior capsulotomy. Severe, rigid contractures in older children may require a distal femoral anterior closing-wedge extension osteotomy.
Foot and Ankle Deformities
The primary goal for the foot in myelomeningocele is to achieve a supple, plantigrade, and braceable foot that is free of pressure ulcerations. Due to insensate skin, any rigid deformity will inevitably lead to skin breakdown, osteomyelitis, and potential amputation.
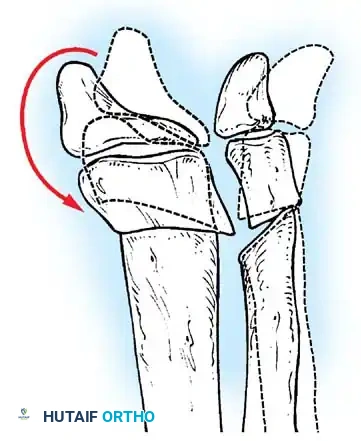
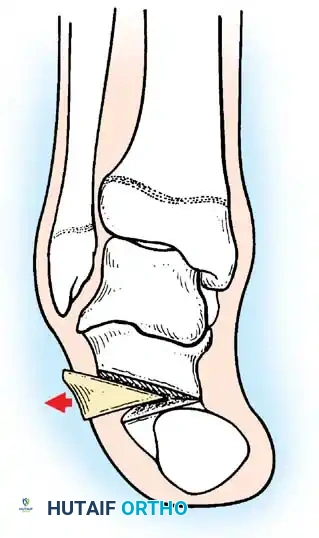
Clubfoot (Talipes Equinovarus)
Clubfoot in myelomeningocele is notoriously rigid and teratologic.
Initial management utilizes the Ponseti method of serial casting. However, the recurrence rate is significantly higher than in idiopathic clubfoot.
When conservative measures fail, surgical intervention is required:
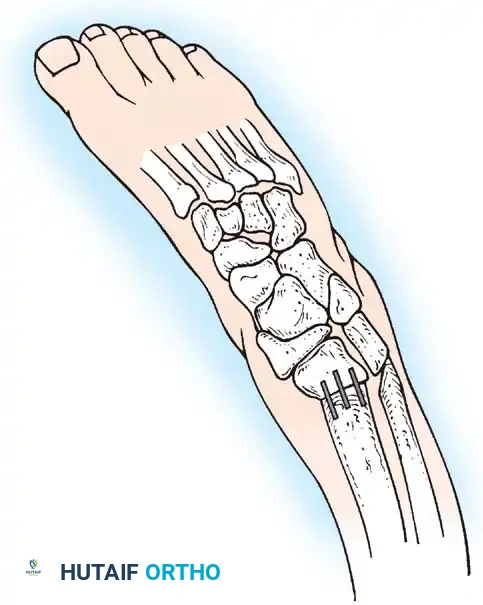
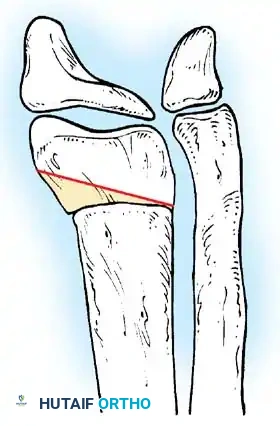
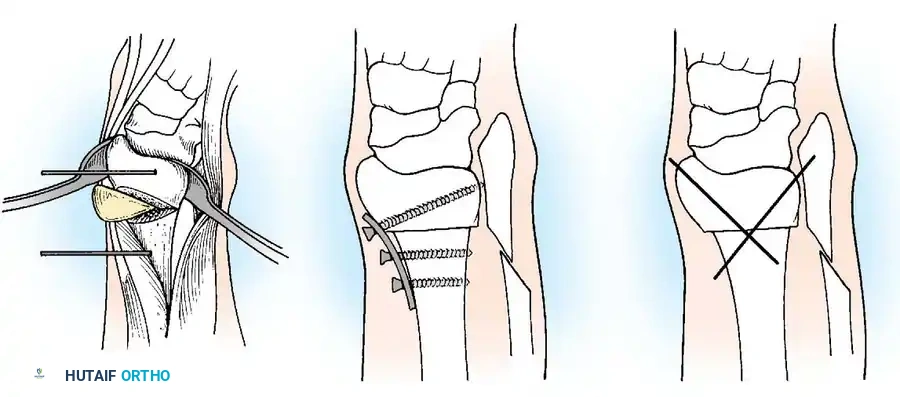
1. Comprehensive Posteromedial Release (PMR): Involves lengthening of the Achilles, posterior tibialis, FDL, and FHL, along with extensive capsulotomies of the ankle, subtalar, and talonavicular joints.

📚 Medical References
- myelomeningocele, Instr Course Lect 28:7, 1979.
- Evans EB, Julian JD: Modifi cations of the hamstring transfer, Dev Med Child Neurol 8:539, 1966.
- Flynn JM, Miller F: Management of hip disorders in patients with cerebral palsy, J Am Acad Orthop Surg 10:198, 2002.
- Fulford GE, Brown JK: Position as a cause of deformity in children with cerebral palsy, Dev Med Child Neurol 18:305, 1976.
- Gabos PG, Miller F, Galban MA, et al: Prosthetic interposition arthroplasty for the palliative treatment of end-stage spastic hip disease in nonambulatory patients with cerebral palsy, J Pediatr Orthop 19:796, 1999.
- Gage JR: Gait analysis for decision making in cerebral palsy, Bull Hosp Jt Dis 43:147, 1983.
- Gage JR, Perry J, Hicks RR, et al: Rectus femoris transfer to improve knee function of children with cerebral palsy, Dev Med Child Neurol 29:159, 1987.
- Gamble JG, Rinsky LA, Bleck EE: Established hip dislocations in children with cerebral palsy, Clin Orthop Relat Res 253:90, 1990.
- Goldner JL: Hip adductor transfer compared with adductor tenotomy in cerebral palsy, J Bone Joint Surg 63A:1498, 1981 (letter). Griffi n PP, Wheelhouse WW, Shiavi R: Adductor transfer for adductor spasticity: clinical and electromyographic gait analysis, Dev Med Child Neurol 19:783, 1977.
- Gross MS, Ibrahim K, Wehner J, et al: Combined surgical procedure for treatment of hip dislocation in cerebral palsy, Orthop Trans 8:113, 1984 (abstract). Gurd AR: Surgical correction of myodesis of the hip in cerebral palsy, Orthop Trans 8:112, 1984 (abstract). Handelsman JE: The Chiari pelvic sliding osteotomy, Orthop Clin North Am 11:105, 1980.
- Hiroshima K, Ono K: Correlation between muscle shortening and derangement of the hip joint in children with spastic cerebral palsy, Clin Orthop Relat Res 144:186, 1979.
- Hodgkinson I, Jindrich ML, Duhaut P, et al: Hip pain in 234 non-ambulatory adolescents and young adults with cerebral palsy: a cross-sectional multicentre study, Dev Med Child Neurol 43:806, 2001.
- Hoffer MM, Prietto C, Koffman M: Supracondylar derotational osteotomy of the femur for internal rotation of the thigh in the cerebral palsied child, J Bone Joint Surg 63A:389, 1981.
- Houkom JA, Roach JW, Wenger DR, et al: Treatment of acquired hip subluxation in cerebral palsy, J Pediatr Orthop 6:285, 1986.
- Howard CB, McKibbin B, Williams LA, et al: Factors affecting the incidence of hip dislocation in cerebral palsy, J Bone Joint Surg 67B:530, 1985.
- Kalen V, Gamble JG: Resection arthroplasty of the hip in paralytic disorders, Dev Med Child Neurol 26:341, 1984.
- Kay RM, Rethlefsen S, Reed M, et al: Changes in pelvic rotation after soft tissue and bony surgery in ambulatory children with cerebral palsy, J Pediatr Orthop 24:278, 2004.
- Keats S: Combined adductor-gracilis tenotomy and selective obturator-nerve resection for the correction of adduction deformity of the hip in children with cerebral palsy, J Bone Joint Surg 39A:1087, 1957.
- Koffman M: Proximal femoral resection or total hip replacement in severely disabled cerebral-spastic patients, Orthop Clin North Am 12:91, 1981.
- Leet AI, Chhor K, Launay F, et al: Femoral head resection for painful hip subluxation in cerebral palsy: is valgus osteotomy in conjunction with femoral head resection preferable to proximal femoral head resection and traction? J Pediatr Orthop 25:70, 2005.
- Letts M, Shapiro L, Mulder K, et al: The windblown hip syndrome in total body cerebral palsy, J Pediatr Orthop 4:55, 1984.
- Lonstein JE, Beck K: Hip dislocation and subluxation in cerebral palsy, J Pediatr Orthop 6:521, 1986.
- Lovejoy SA, Tylkowski C, Oeffi nger D, et al: The effects of hamstring lengthening on hip rotation, J Pediatr Orthop 27:142, 2007.
- Lubicky JP, Bernotas S, Herman JE: Complications related to postoperative casting after surgical treatment of subluxed/dislocated hips in patients with cerebral palsy, Orthopedics 26:407, 2003.
- Lundy DW, Ganey TM, Ogden JA, et al: Pathologic morphology of the dislocated proximal femur in children with cerebral palsy, J Pediatr Orthop 18:528, 1998.
- Lynne ED, Katcherian DA: Slotted acetabular augmentation in patients with
You Might Also Like