Understanding Benign Bone Tumors: Your Complete Preoperative Guide
Key Takeaway
For anyone wondering about Understanding Benign Bone Tumors: Your Complete Preoperative Guide, Bone tumors encompass both benign bone tumors and malignant sarcomas. Bone sarcomas are rare, aggressive malignancies originating from mesenchymal tissue, representing less than 0.2% of adult cancers. Types include osteosarcoma and chondrosarcoma. Understanding their unique biologic behavior is essential for appropriate staging and treatment planning, influencing the use of adjuvant therapies.
Introduction and Epidemiology of Musculoskeletal Tumors
An exhaustive understanding of the basic biology, genetics, and histopathology of bone and soft tissue tumors is an absolute prerequisite for the appropriate staging, preoperative planning, and surgical execution of oncologic resections. This comprehensive review delineates the unique biologic behavior of soft tissue sarcomas (STS) and primary bone sarcomas. These behavioral patterns provide the foundational rationale for current staging systems, the determination of surgical margins, and the integration of neoadjuvant and adjuvant treatment modalities.
Soft tissue and bone sarcomas represent a rare, highly heterogeneous group of mesenchymal neoplasms. Epidemiologically, these tumors account for less than 0.2% of all adult malignancies, yet they represent a disproportionately high 15% of pediatric malignancies, emphasizing the critical need for specialized pediatric and orthopedic oncologic care. The annual incidence in the United States remains relatively stable, with approximately 11,400 new cases of soft tissue sarcomas and 3,010 new cases of primary bone sarcomas diagnosed annually. Mortality data reflects the aggressive nature of these diseases, with an estimated 4,390 deaths attributed to STS and 1,440 deaths to bone cancers each year.
Incidence and Prevalence by Age and Histology
The distribution of primary bone cancer histology is highly age-dependent. In the adult population, chondrosarcomas predominate, accounting for over 40% of primary bone malignancies. This is followed by osteosarcoma (OS) at 28%, chordomas (10%), Ewing sarcoma (8%), and undifferentiated pleomorphic sarcoma (UPS)/fibrosarcoma (4%). The remainder consists of exceedingly rare osseous malignancies such as adamantinoma and angiosarcoma of bone.
Conversely, in the pediatric and adolescent demographic (patients younger than 20 years), osteosarcoma is the most prevalent primary bone tumor (56%), followed by Ewing sarcoma (34%). Chondrosarcoma is distinctly rare in this cohort, representing only 6% of cases.
Survival Rates and Prognostic Trends
Advancements in multimodality therapy have significantly improved survival trajectories. In the United States, the overall 5-year survival rate for all primary bone sarcomas is approximately 70%. For adolescents and young adults (15 to 29 years old), osteosarcoma and Ewing sarcoma demonstrate comparable 5-year survival rates of approximately 60% in the modern chemotherapeutic era. Mortality from bone cancer peaks in the 15 to 19-year age group, correlating with the incidence spikes of OS and Ewing sarcoma during the adolescent growth spurt. For soft tissue sarcomas, the 5-year survival rate for localized disease is highly favorable at 83%; however, the presence of systemic metastasis precipitates a precipitous drop in survival to 16%.
Risk Factors and Genetic Syndromes
While the majority of musculoskeletal sarcomas arise sporadically without a discernible etiology, several distinct environmental, iatrogenic, and genetic risk factors have been identified. Recognizing these risk factors is essential for appropriate patient screening and genetic counseling.
- Environmental and Iatrogenic Exposures: Prior therapeutic radiation is a well-documented risk factor, often leading to secondary osteosarcoma, UPS, or angiosarcoma within the radiation field years or decades later. Chemical exposures (e.g., vinyl chloride linked to hepatic angiosarcoma, arsenic) and chronic immunosuppression are also implicated.
- Chronic Tissue Irritation: Sarcomas can arise in the setting of chronic inflammation, including prior severe injuries (burn scars leading to Marjolin's ulcer or sarcomatous change), chronic lymphedema (Stewart-Treves syndrome), foreign body implants, and chronic osteomyelitis.
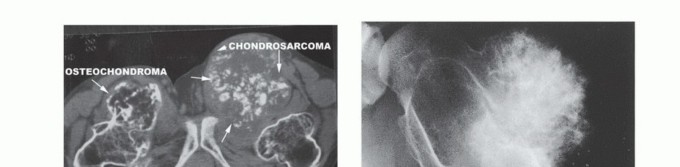
- Pre-existing Benign Bone Conditions: Paget disease of bone is a classic precursor for secondary osteosarcoma in the elderly. Bone infarcts, fibrous dysplasia, and osteochondromas also carry a risk of malignant transformation.
- Genetic Cancer Syndromes: A thorough family history is paramount. Key syndromes include:
- Li-Fraumeni Syndrome: Germline p53 mutation, highly associated with osteosarcoma and STS.
- Hereditary Retinoblastoma: RB1 mutation, exponentially increasing the risk of secondary osteosarcoma.
- Familial Adenomatous Polyposis (Gardner Syndrome): Associated with aggressive fibromatosis (desmoid tumors) and osteomas.
- Other DNA Repair/Helicase Defects: Rothmund-Thomson syndrome, Werner syndrome, and Bloom syndrome.
- Cartilage Syndromes: Maffucci syndrome and Ollier disease (enchondromatosis), and Hereditary Multiple Exostoses (EXT1/EXT2 mutations), which predispose patients to secondary chondrosarcomas.
Pathophysiology and Biologic Behavior
Sarcomas originate primarily from the embryonic mesoderm. Soft tissue sarcomas are broadly classified based on the mature tissue they most closely resemble histologically (e.g., liposarcoma resembles adipose tissue, leiomyosarcoma resembles smooth muscle). Bone sarcomas are classified by the dominant extracellular matrix produced by the malignant cells: osteoid-producing tumors are osteosarcomas, while chondroid-producing tumors are chondrosarcomas.
The biologic behavior of these tumors is dictated by their shared mesenchymal origin and their specific anatomic microenvironment. These unique growth patterns form the foundational logic for surgical staging and compartmental resection strategies.
Classification and Histologic Grading
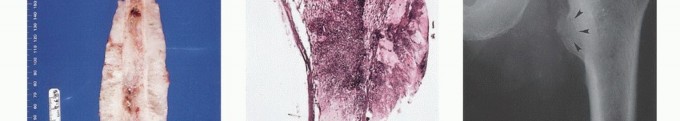
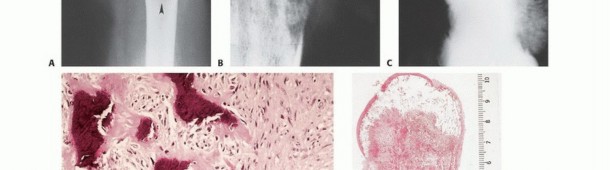
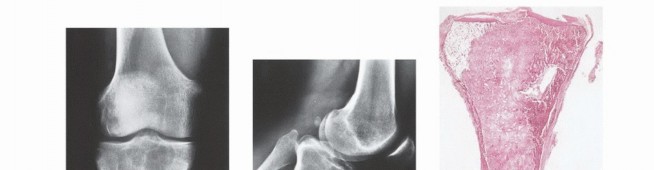
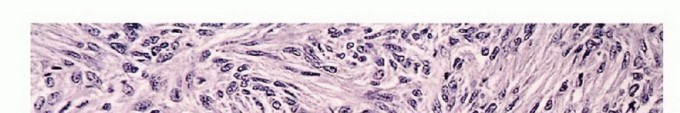
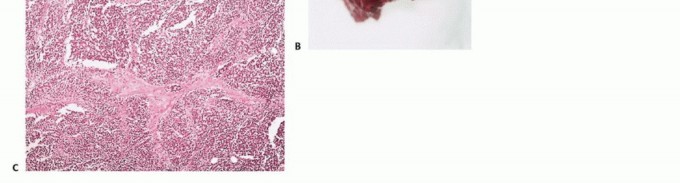
Histologic grading is the most critical prognostic indicator for sarcomas. Tumors are graded as low, intermediate, or high grade. The assignment of grade depends on a composite assessment of tumor morphology, cellular pleomorphism, nuclear atypia, matrix production, and most importantly, mitotic index and the presence of spontaneous tumor necrosis.
Tumor grade directly reflects the biologic aggressiveness and the statistical probability of systemic metastasis. Low-grade lesions are locally aggressive but rarely metastasize. High-grade lesions possess a high metastatic potential, disseminating in over 20% of patients, and often present with occult micrometastases.
Tumor Growth Dynamics and the Pseudocapsule
Unlike carcinomas, which grow via direct infiltrative destruction, sarcomas typically form a solid, cohesive mass that expands centrifugally. The periphery of the lesion is the most biologically active and least mature zone.
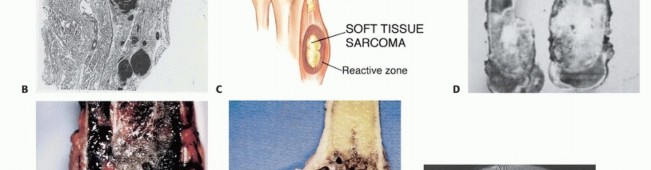
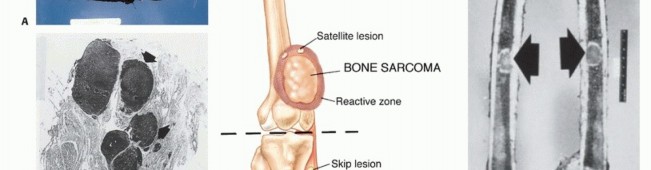
Benign lesions are often constrained by a true capsule composed of compressed, mature normal cells. In stark contrast, sarcomas are enveloped by a "pseudocapsule." This reactive zone consists of compressed malignant cells, a fibrovascular stroma, and a dense inflammatory infiltrate interacting with the surrounding host tissue. The pseudocapsule is highly deceptive; it is not a true anatomic barrier but rather a tumor-host interface.
The thickness and integrity of this reactive zone vary with tumor grade. High-grade sarcomas feature a poorly defined, highly permeable reactive zone that is frequently invaded by microscopic tumor extensions.
* Satellite Lesions: Microscopic or macroscopic tumor foci located within the reactive zone/pseudocapsule.
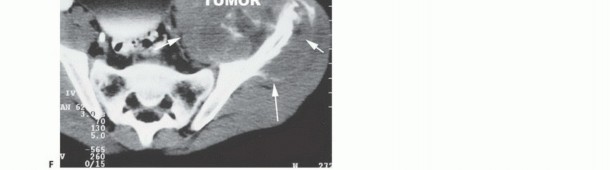
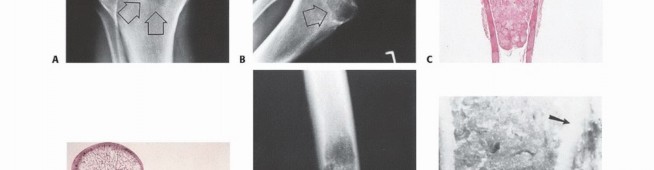
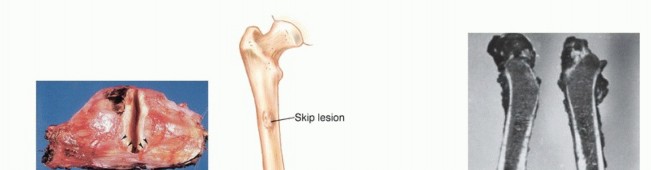
* Skip Metastases: Tumor nodules that have broken through the pseudocapsule and reside outside the reactive zone but remain within the same anatomic compartment. These represent locoregional micrometastases that have spread via local tissue planes or marrow sinusoids, independent of systemic circulation. The presence of skip metastases (seen in <5% of osteosarcomas preoperatively) is a dire prognostic sign and accounts for local recurrences when seemingly adequate margins are achieved without recognizing the skip lesion. Low-grade sarcomas may interdigitate into the reactive zone but rarely form true skip metastases.
Anatomic Barriers and Compartmentalization
Sarcomas generally respect major anatomic boundaries, taking the path of least mechanical resistance. They initially expand within the specific anatomic compartment of origin (e.g., the anterior compartment of the thigh, or the medullary canal of a bone). Only in advanced stages, or following iatrogenic violation (e.g., a poorly planned biopsy), do they breach the compartment walls—such as thick muscular aponeuroses, fascia, or cortical bone—to invade adjacent compartments.
- Effective Barriers: Articular cartilage, dense cortical bone, and thick fascial septae are robust natural barriers to tumor extension.
- Ineffective Barriers: The pediatric growth plate (physis) is not a reliable barrier. The rich transphyseal vascular channels present during growth allow tumor to readily pass from the metaphysis into the epiphysis.
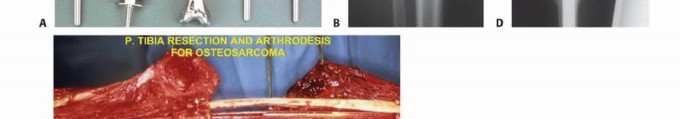
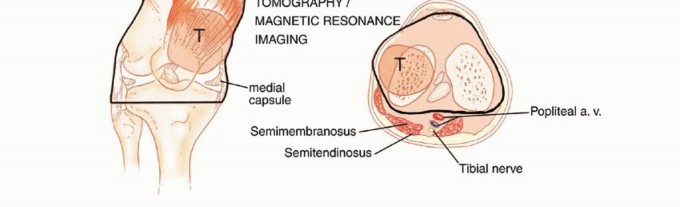
Tumors are defined surgically as intracompartmental if they are entirely contained within their native anatomic compartment. They are extracompartmental if they have breached a fascial/cortical barrier, or if they originate in an uncompartmentalized space (e.g., the popliteal fossa, axilla, groin, or antecubital fossa). Most high-grade primary bone sarcomas are bicompartmental at presentation, having destroyed the native cortex via the Haversian and Volkmann canals to form a contiguous soft tissue mass. Carcinomas metastatic to the extremity, unlike primary sarcomas, ignore compartmental boundaries and exhibit true infiltrative growth.
Mechanisms of Joint Involvement
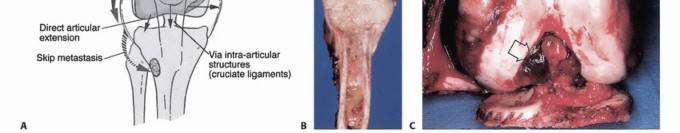
Direct transarticular extension of a sarcoma through intact articular cartilage is exceedingly rare. When a joint is involved, it typically occurs via one of the following mechanisms:
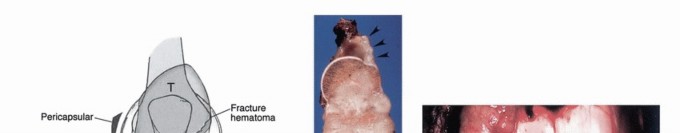
1. Pathologic fracture extending into the intra-articular space, seeding the joint with tumor cells.
2. Pericapsular extension, where the soft tissue component of the tumor wraps around the joint capsule and invades the synovium.
3. Extension along intra-articular structures that insert directly into the bone (e.g., tumor tracking along the cruciate ligaments into the knee).
4. Transcapsular skip metastases (documented in ~1% of osteosarcomas).
Metastatic Dissemination
The dissemination of bone and soft tissue sarcomas occurs almost exclusively via the hematogenous route. This is a critical distinction from carcinomas and melanomas, which preferentially utilize the lymphatic system. Hematogenous spread of extremity sarcomas most frequently manifests as pulmonary metastases, followed by secondary osseous metastases. Sarcomas arising in the abdominal or pelvic retroperitoneum typically metastasize to the liver and lungs via the portal and systemic venous systems.
Lymphatic spread to regional nodes is rare, occurring in approximately 13% of STS (notably synovial sarcoma, clear cell sarcoma, epithelioid sarcoma, rhabdomyosarcoma, and angiosarcoma) and only 7% of bone sarcomas.
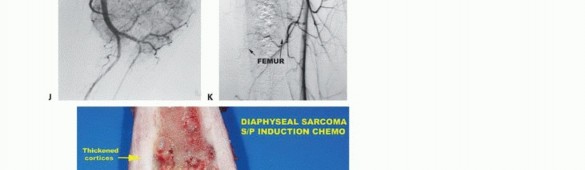
Because high-grade bone sarcomas (like OS and Ewing) have a profound propensity for early hematogenous dissemination, it is estimated that 80% of patients have occult pulmonary micrometastases at the time of initial diagnosis. Consequently, surgical resection alone is insufficient for cure; systemic neoadjuvant and adjuvant chemotherapy are mandatory.
Oncologic Staging Systems
Staging is the systematic classification of a neoplasm based on its histologic grade, local anatomic extent, and the presence of systemic dissemination. Accurate staging dictates the surgical approach, the need for systemic therapy, and establishes the prognosis. Unlike staging for carcinomas, which relies heavily on lymph node status (TNM), the most critical variable in musculoskeletal sarcoma staging is the histologic grade.
AJCC Staging System
The American Joint Committee on Cancer (AJCC) staging system is widely utilized for soft tissue sarcomas and bone sarcomas. For STS, it evaluates Primary Tumor Size and Depth (T1/T2, superficial vs. deep), Regional Lymph Nodes (N), Distant Metastasis (M), and Histologic Grade (G1-G3). A deep tumor is defined as one located beneath the superficial fascia. Any presence of nodal metastasis (N1) automatically elevates the staging to Stage III, reflecting a prognosis similar to distant metastasis.
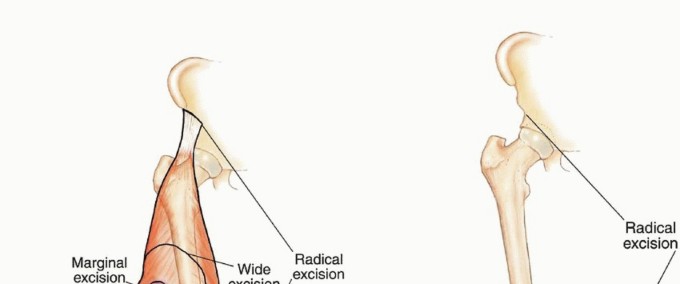
Enneking Surgical Staging System for Malignant Tumors
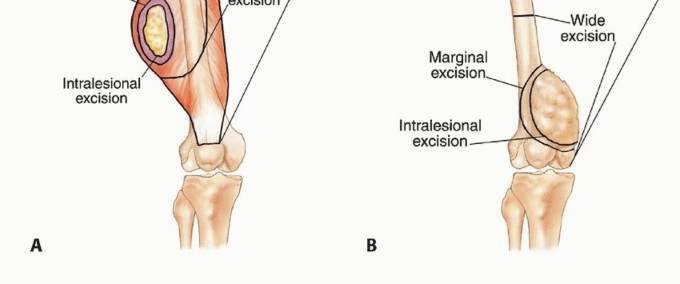
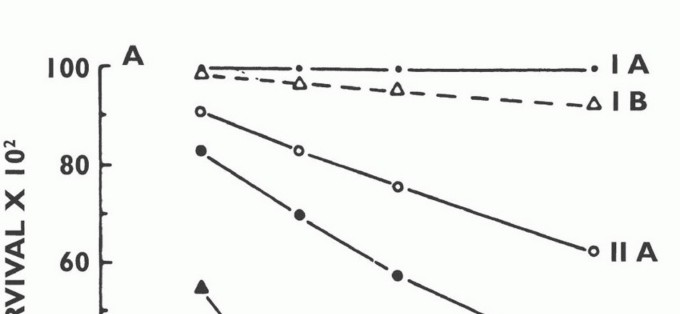
Adopted by the Musculoskeletal Tumor Society (MSTS), the Enneking system is elegantly tailored to the surgical management of sarcomas. It relies on three pillars: Histologic Grade (G1 = Low, G2 = High), Anatomic Site (T1 = Intracompartmental, T2 = Extracompartmental), and Metastasis (M0 = None, M1 = Regional/Distant).
- Stage IA: Low Grade, Intracompartmental (G1, T1, M0)
- Stage IB: Low Grade, Extracompartmental (G1, T2, M0)
- Stage IIA: High Grade, Intracompartmental (G2, T1, M0)
- Stage IIB: High Grade, Extracompartmental (G2, T2, M0)
- Stage III: Any Grade, Any Site, Metastatic (Any G, Any T, M1)
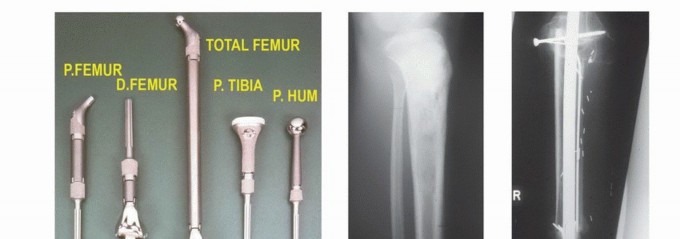
Developed before the widespread use of neoadjuvant chemotherapy, this system directly correlated the anatomic extent of the tumor with the necessary surgical margin. While modern neoadjuvant therapy often shrinks the soft tissue component and facilitates limb salvage over amputation, the Enneking system remains the gold standard for understanding the biologic behavior and surgical anatomy of the lesion.
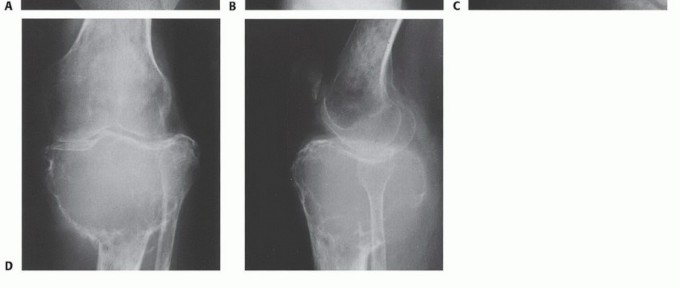
Enneking Staging System for Benign Bone Tumors
Enneking also established a robust staging system for benign osseous lesions based on clinical presentation and radiographic behavior:
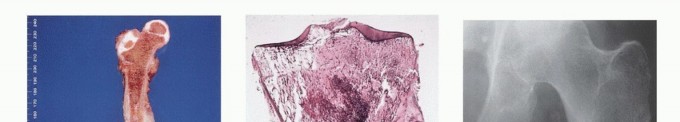
* Stage 1 (Latent): Asymptomatic, discovered incidentally. Static growth or spontaneous healing. Thick, well-defined reactive sclerotic rim. Treated with observation or simple curettage (e.g., Non-ossifying fibroma).
* Stage 2 (Active): Progressive growth, mild symptoms, but contained within natural cortical barriers. Treated with extended curettage and burr drilling (e.g., Aneurysmal bone cyst).
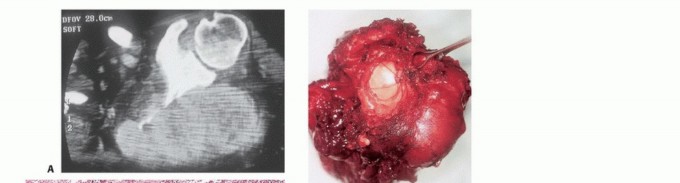
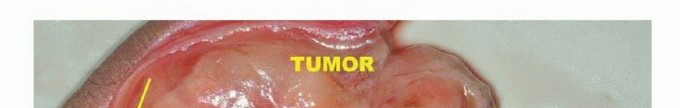
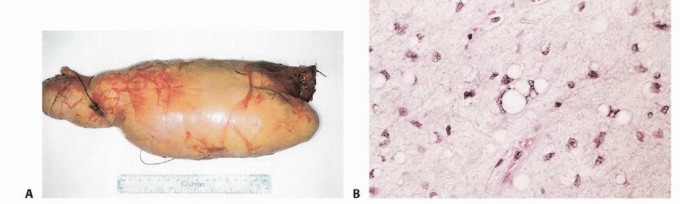
* Stage 3 (Aggressive): Rapid growth, symptomatic, destroys cortical bone, and extends into soft tissue. Minimal to no reactive bone rim. Requires aggressive extended curettage with local adjuvants (liquid nitrogen, phenol, argon beam) or wide en bloc resection (e.g., Giant cell tumor of bone).
Imaging Set 1: Pathophysiology and Staging Fundamentals


Clinical Evaluation and Diagnostic Imaging
Presenting Symptoms and Physical Examination
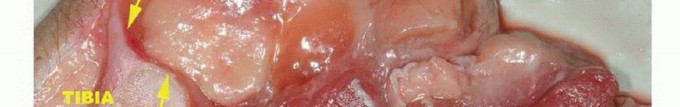
The clinical presentation of bone sarcomas is classically characterized by deep, dull, aching pain that mimics a toothache. The pain is initially intermittent, often associated with activity, but progresses to constant, unremitting pain, notably including night pain that awakens the patient. High-grade tumors typically present with a rapid onset of symptoms (weeks to months), whereas low-grade lesions may cause indolent pain over many months. Localized soft tissue swelling, a palpable mass fixed to the underlying bone, and localized erythema or venous engorgement may be present. Pathologic fractures occur in a minority of cases but complicate management significantly.
Soft tissue sarcomas most frequently arise in the lower extremities (46%), followed by the trunk (19%), upper extremities (13%), and retroperitoneum (12%). They typically present as a painless, progressively enlarging mass. However, in approximately 20% of cases, rapid expansion, hemorrhage, or nerve compression results in a painful presentation. A comprehensive physical examination must assess the mass size, depth, mobility, overlying skin changes, distal neurovascular status, and regional lymphadenopathy.
Formulating an Initial Assessment
The formulation of a differential diagnosis in orthopedic oncology relies heavily on patient age, anatomic location, and radiographic appearance.
* Age:
* 0-10 years: Ewing sarcoma, leukemia, metastatic neuroblastoma.
* 10-25 years: Osteosarcoma, Ewing sarcoma, osteochondroma, non-ossifying fibroma.
* >40 years: Metastatic carcinoma, multiple myeloma, chondrosarcoma, secondary osteosarcoma (Paget's), lymphoma.
* Location:
* Epiphysis: Chondroblastoma, Giant Cell Tumor (after physeal closure).
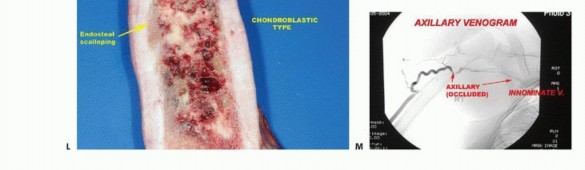
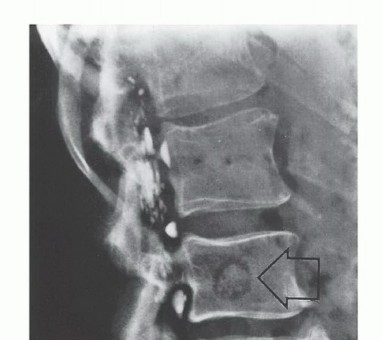
* Metaphysis: Osteosarcoma, Chondrosarcoma.
* Diaphysis: Ewing sarcoma, Adamantinoma (tibia), Lymphoma.
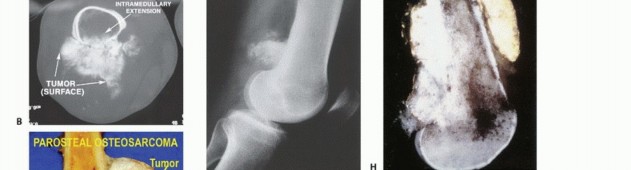
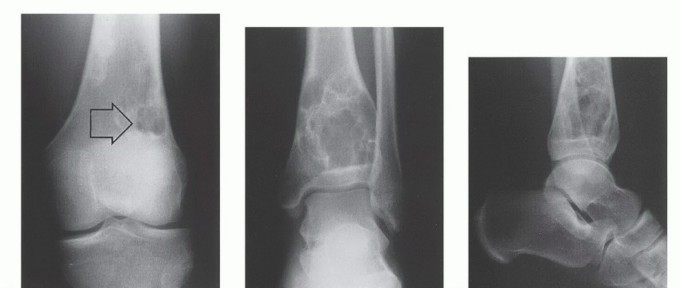
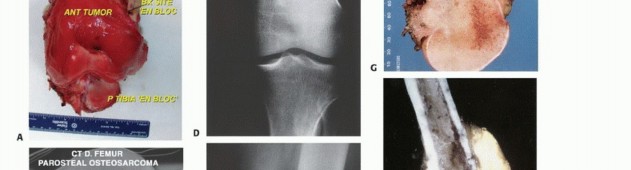
* Surface: Parosteal/Periosteal osteosarcoma.
* Matrix Production:
* Osteoid: Cloud-like, amorphous sclerosis.
* Chondroid: Stippled, punctate, "popcorn" calcifications.
* Fibrous: Ground-glass appearance.
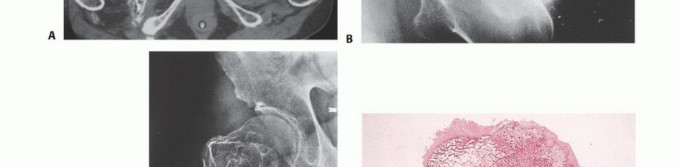
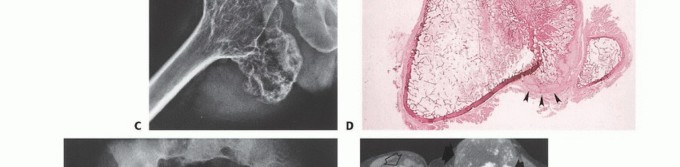
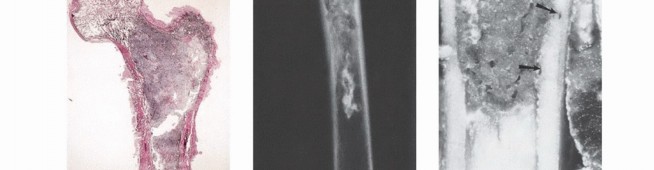
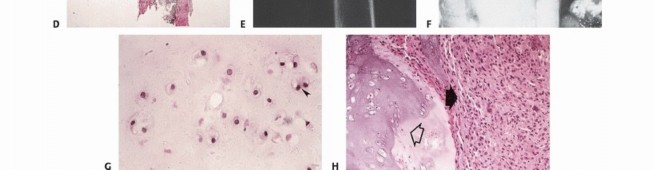
Radiographic and Advanced Imaging Modalities
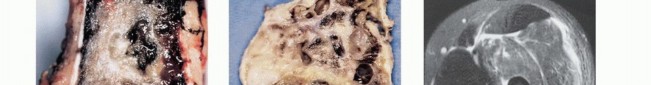
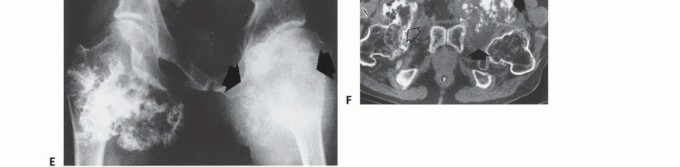
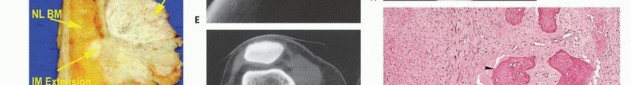
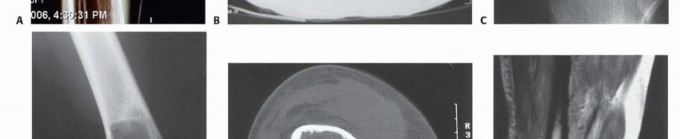
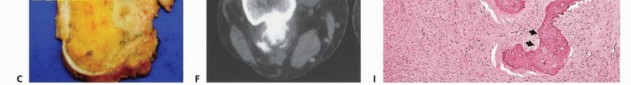
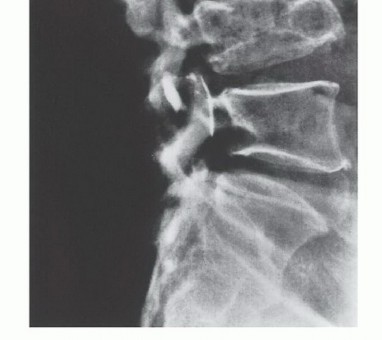
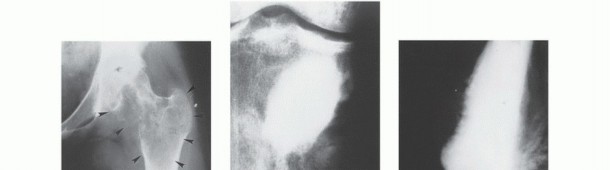
- Plain Radiography: The cornerstone of initial evaluation. It accurately predicts the diagnosis in over 80% of extremity bone tumors. It defines the lesion's location, zone of transition (narrow/sclerotic = benign/slow; wide/permeative = malignant/aggressive), cortical destruction, and periosteal reactions (Codman triangle, onion-skinning, sunburst pattern).
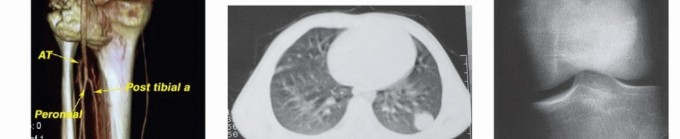
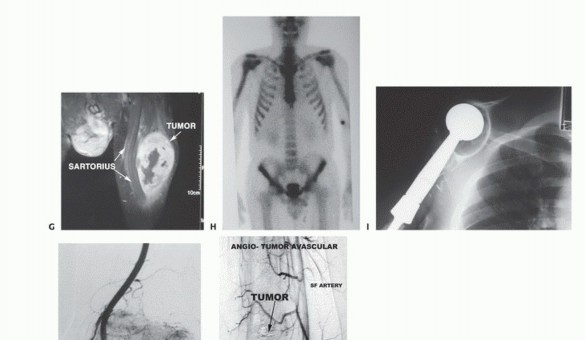
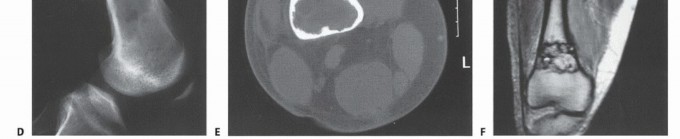
- Computed Tomography (CT): The modality of choice for assessing fine osseous detail, cortical integrity, and matrix mineralization. Thin-slice (≤1 mm) helical CT allows for precise 3D reconstructions. Intravenous contrast is essential to delineate the relationship of the soft tissue mass to major vascular bundles. Chest CT is mandatory for staging to rule out pulmonary metastases.
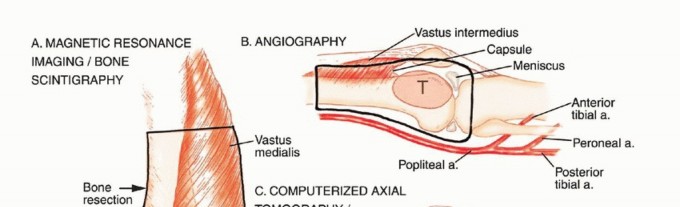
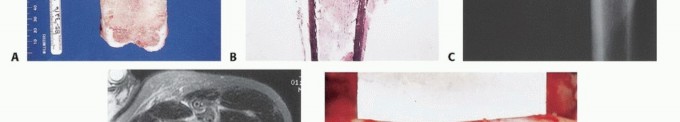
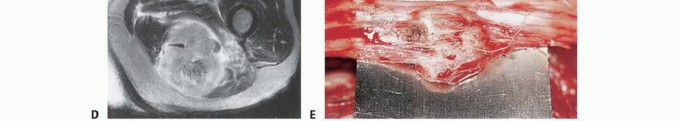
- Magnetic Resonance Imaging (MRI): The gold standard for evaluating the local extent of the tumor. It accurately defines the intramedullary extent (allowing calculation of bone resection levels), soft tissue extension, joint involvement, and the presence of skip metastases. T1-weighted images best define marrow replacement; T2-weighted and STIR sequences highlight peritumoral edema and the soft tissue mass. Contrast enhancement differentiates cystic from solid components and clarifies neurovascular proximity.
- Bone Scintigraphy (Technetium-99m): Utilized to detect polyostotic disease and skeletal metastases. The three-phase bone scan assesses biologic activity; a "tumor blush" in the flow/pool phases indicates high vascularity typical of malignancy.
- Angiography and Venography: While largely supplanted by CT/MR Angiography, traditional angiography can map complex vascular displacements, identify feeding vessels for preoperative embolization (critical for hypervascular metastases like renal cell carcinoma), and assess the patency of collateral circulation prior to major vessel ligation. Venography can identify tumor thrombus or extrinsic venous compression.
- PET-CT (FDG): A functional imaging modality measuring glucose metabolism. It is increasingly the standard of care for total body staging, identifying occult metastases, and monitoring the metabolic response to neoadjuvant chemotherapy (measured by the Standardized Uptake Value, SUV).
Laboratory Studies and Biopsy
Routine laboratory studies (CBC, ESR, CMP) are often non-specific but necessary. In patients over 40, serum and urine electrophoresis (SPEP/UPEP) are required to rule out multiple myeloma. In osteosarcoma, elevated serum alkaline phosphatase and lactate dehydrogenase (LDH) are independent negative prognostic indicators.
Biopsy is the final, definitive step in staging. It must be meticulously planned by the treating orthopedic oncologist. Poorly executed biopsies can contaminate tissue planes, necessitating amputation instead of limb salvage. Core needle biopsy is preferred; if open incisional biopsy is required, it must be longitudinal, meticulously hemostatic, and placed entirely within the planned definitive resection tract.
Imaging Set 2: Advanced Diagnostic Modalities










You Might Also Like